
Multi-dimensional correlation of layered Li-rich Mn-based cathode materials

Abstract
Lithium-rich manganese-based cathode materials are expected to promote the commercialization of lithium-ion batteries to a new stage by virtue of their ultrahigh specific capacity and energy density advantages. However, they are still restricted by complex phase transitions and electrochemical performance degradation caused by labile anion charge compensation. A deep understanding of the electrochemical properties contained in their intrinsic structures and the key driving factors of structural deterioration during cycling are crucial to guide the preparation and optimization of lithium-rich materials. Considering recent progress, this review introduces the intrinsic properties of Li-rich manganese-based cathode materials from interatomic interactions to particle morphology at multiple scales in the spatial dimension. The charge compensation mechanism and energy band reorganization of the initial charge and discharge, the structural evolution during cycling and the electrochemical reaction kinetics of the materials are analyzed in the temporal dimension. Based on the relationship between structure and electrochemical performance, preparation methods and modification methods are introduced to guide and design cathode materials. Effective characterization methods for studying anion charge compensation behavior are also demonstrated. This review provides important guidance and suggestions for making full use of the high specific capacity in these materials derived from anion redox and the maintaining of its stability.
Keywords
INTRODUCTION
The energy revolution is in the ascendancy. With the current global climate deteriorating, the reduction of carbon emissions has become an important measure to address climate change[1]. The main measures to reduce carbon emissions are to reduce the use of fossil energy and vigorously develop sustainable clean energy. As state-of-the-art electrochemical energy storage and conversion devices, lithium-ion batteries (LIBs) have been widely used in all aspects of modern society, including 3C products, electric vehicles and energy storage, and are an important link in achieving energy optimization and transformation[2].
High energy density is the eternal development direction for LIBs. From the 80 Wh kg-1 energy density of the first generation of commercial LIBs launched by Sony in the 1990s, LIBs have gone through several innovations in every aspect, from materials to assembly[3]. Cathode materials account for the largest cost and proportion of LIBs and are therefore significant limiting factors in reducing costs and increasing energy density. Current commercial cathode materials, including layered LiCoO2, LiNixCoyMn1-x-yO2 (NCM) and LiNixCoyAl1-x-yO2, spinel LiMn2O4 and olivine LiFePO4[4], have gradually reached their theoretical capacity energy density. In order to break through the energy bottleneck of LIBs, electrode materials need to be reformed in the electrochemical reaction system. Compared with the traditional cationic charge compensation mechanism, the unique anion redox mechanism of lithium-rich manganese-based layered oxides (LRMOs), xLiMO2·(1-x)Li2MnO3 (M = Ni, Co, Mn, Ni1/2Mn1/2, Ni1/3Co1/3Mn1/3 and so on), creates conditions for achieving higher specific capacity and energy density. These materials can achieve a high specific capacity exceeding 250 mAh g-1 in a large voltage range of 2.0-4.8 V, thereby making them ideal candidates for the next generation of LIB cathode materials[5].
Nevertheless, LRMOs still face a series of challenges that seriously hindered their commercialization. These challenges include: (1) a lack of systematic understanding in their intrinsic structure-activity relationships at multiple spatial scales; (2) low Coulombic efficiency and serious voltage hysteresis caused by the violent structural reorganization during the initial charge-discharge cycle; (3) capacity attenuation and voltage decay due to phase transformations and the dissolution of active Mn with deposition on the anode electrode[6,7]; (4) poor rate performance limited by sluggish electrode reaction kinetics[8]; and (5) reasonable and efficient materials preparation, modification and characterization methods that require substantial organization and improvement.
With extensive surveying and the employment of more advanced atomic-scale in-situ/ex-situ characterization technologies, the in-depth mechanisms behind the above-mentioned problems and their optimization/modification have achieved remarkable progress. Based on research of model materials, such as Li2MO3 (M = Mn[9], Ru[10], Ir[11], Mo[12] and Ti[13]), the trigger mechanism and reversibility of anion redox and its influence on the structure and electrochemical performance have been gradually clarified. For LRMO cathodes, the monolithic materials undergo a series of complex phase evolutions during electrochemical cycling, including irreversible migration of in-plane/out-of-plane transition metals (TMs)[14], evolution of the superlattice structure, generation and diffusion of oxygen vacancies, lattice structure distortion and deterioration and the continuous transition of redox couples[15]. In addition, more serious O2 release, element dissolution, electrolyte erosion and structure densification occur in the near-surface interlayer of the material particles. In view of the above, surface modification[16], bulk doping[17], morphology control[18], element ratio[19], local structure[20], electrode/electrolyte[21] and so on, have been considered to improve the structural stability and electrochemical performance of these cathode materials from different perspectives.
Based on the traditional view and recent research progress into LRMOs[22-24], this review elaborates the intrinsic structure, electrochemical properties and their relationship in the scope of multiple spatial scales (atomic scale, local structure, particle morphology and so on). The temporal clues of these materials in electrochemical processes also demonstrate the structure and energy band changes that cause inconsistency in the initial charge-discharge behavior, the influence of the local structural evolution on the electrochemical performance during cycling and the restrictive factors of the sluggish electrode reaction kinetics. The effective means for improving the electrochemical performance of LRMOs are reported from the aspects of materials preparation and modification. Classical electrochemistry and advanced physical characterization methods are introduced to comprehensively study the electrochemical reaction, structural evolution and anion charge compensation mechanism of LRMOs. Finally, various challenges that still exist in current research, countermeasures and future research directions are proposed (Scheme 1).
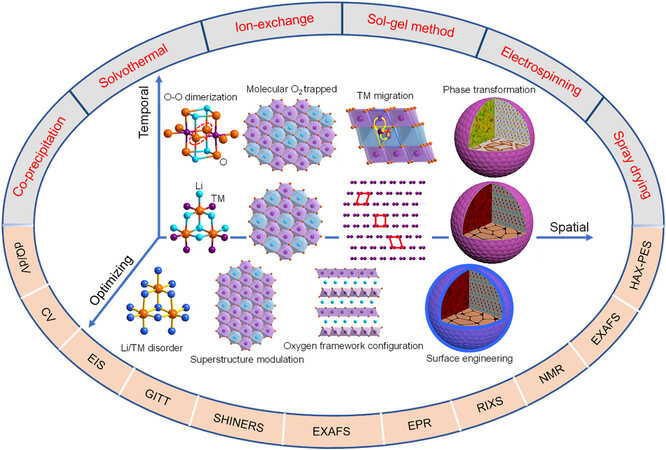
Scheme 1. Summary of layered Li-rich Mn-based cathode materials in multiple dimensions.
SPATIAL DIMENSION
The derivation of cathode performance depends on its structure and the understanding of structure-activity relationships is an ever-important topic. In order to clarify the electrochemical characteristics and the evolution laws of lithium-rich cathode materials, it is necessary to consider them from a multiscale perspective with regards to electron/energy band configuration, local microstructure and particle morphology.
Spatial coordination of elements
Li-O-Li configuration
LRMO cathode materials consist of LiMO2 (M = Ni, Co, Mn and so on) and Li2MnO3 components, where Li and TM atoms are located at the center of octahedra surrounded by six oxygen atoms and the atom layer alternately arranges as O/Li/O/TM along the c axis. The occupation of extra Li atoms at the TM sites results in a √3a × 3a LiMn6 superstructure, which gives rise to a change in the coordination environment of oxygen, i.e., three Li-O-TM configurations transform into two Li-O-TM configurations and a special Li-O-Li configuration[25] (shown in Figure 1A). The intense interaction of Li-O ionic bonds leaves the distribution of coordination electrons between Li and O closer to the oxygen element, forming an isolated unhybridized O 2p orbital. The Li-O-Li configurations stimulate a higher energy band, enabling the charge compensation of oxygen during charging (the downward shift of the Fermi level). TM-O bonds rotate along the other directions, resulting in the coplanar arrangement of adjacent unhybridized O 2p orbitals, which interact with each other to ultimately form O-O dimers. The unhybridized oxygen orbitals caused by the Li-O-Li configurations can also be triggered by other configurations[26] (such as Li-O-vacancy and Li-O-Na configurations).
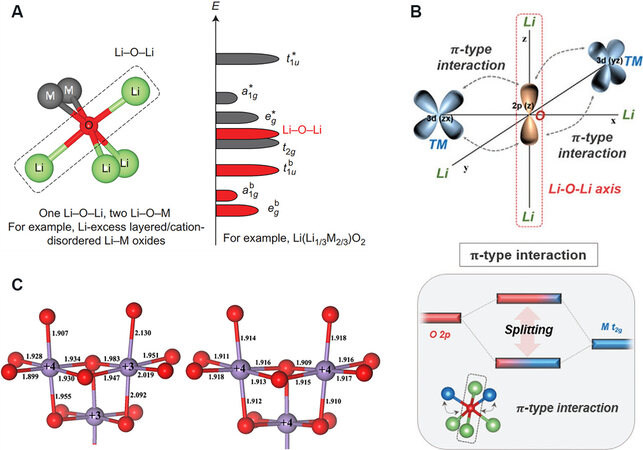
Figure 1. (A) Local coordination of oxygen with one Li-O-Li and two Li-O-M configurations and a schematic of the band structure for Li-rich layered oxides[25]. (B) Schematic of π-type hybridization of the Li-O-Li axis with neighboring TM and the band structure for the π-type hybridized state of the Li-O-Li axis interacted with M t2g[27]. (c) Local coordination around Mn3+ and Mn4+ in LiMn2O4 (left) and λ-MnO2 (right) obtained from theoretical calculations. The gray and red spheres represent Mn and oxygen atoms, respectively[33].
TM-O hybridization
The charge compensation of anions is more closely associated with TMs coordinated by oxygen atoms[27], which is attributed to the two Li-O-TM configurations and one Li-O-Li configuration. For the typical Mn-based material Li2MnO3, Mn bonds with oxygen in a form where the non-bonding oxygen energy band with high activity is separated from the Mn t2g energy band and electrons directly escape from it, contributing to the severe irreversible release of oxygen[28]. The introduction of Fe into the inert structure of Li2TiO3 successfully motivates the anion redox activity[13]. This can be attributed to the fact that electron holes tend to be selectively generated on oxygen ions bonded to Fe atoms instead of Ti atoms. The mechanism can also be extended to the stimulation of anions in Co-doped Li2TiS3.
In the model Li2RuO3 material, the strong covalent interaction between Ru and oxygen causes overlap of the Ru 4d and O 2p non-bonding bands, exhibiting a competitive anion-cation redox coupling reaction with highly reversible anion redox[10]. The redox activity of Li2RuO3 can be regulated by the weaker covalency of the Sn-O bond. Furthermore, the intensely bonding Ir-O immensely suppresses the charge compensation ability of oxygen for Li2IrO3 and all the capacity derives from the redox reaction of Ir hybridized with oxygen[29]. When Ir is partially replaced by Sn, the lower number of valence electrons at the end of charging significantly promotes the ligand-to-metal charge transfer, resulting in short Ir-O π bonds and O-O dimers with a bond length of 1.4 Å. In Li2RhO3, with an intense π-type interaction [Figure 1B], the weaker difference in electronegativity between Rh and oxygen causes a more intense TM-O interaction and increases the contribution of O 2p to the high-energy π* orbital. Oxygen exhibits high reactivity under the occupied anti-bonding state as the π electron donor of the central Rh, displaying a novel oxygen redox reaction at low voltages[30] (which is different from the redox of non-bonding oxygen), where oxygen participates in the reaction prior to Rh. The functional mechanism is demonstrated to be triggered by the oxygen-dominated
As a result, the non-bonding O energy band is subtly regulated by the varieties and numbers of adjacent TM elements, owing to the π-type interaction between Li-O-Li configuration and TM t2g bands. The hybridization state of TM-O is key for adjusting the anion charge compensation ability. The ability to resist oxygen loss and structural deterioration is promoted with the increase in M-O covalent bonds[31]. The redox mechanism derived from the 3d metals and more heavy metals with a d0 electron configuration in Li2MnO3 is related with the lone-pair electron consumption of oxygen (locally non-bonding O 2p state), which leads to irreversible anion redox. Electrode materials formed by the coordination hybridization between oxygen and 4d and 5d metals with intermediate dn electron configurations are able to induce reversible cation and anion redox activity to realize reversible extra capacity.
The anion and cation redox ability can be visibly judged according to a Zaanen-Sawatzky-Allen diagram via the comparison of U (Coulombic interaction, depending on the d metal) and D (energy difference between (M-O) and (M-O)*, known as the charge transfer parameter)[32]. When U << D, the d band established in the (MO)* state locates first on the Fermi level, resulting in pure cation redox. When U > > D, the O 2p state on the p band lies at the Fermi level, thereby motivating anion redox that leads to the generation of highly unstable electron holes and highly activated free radicals in localized states. O-O dimers then form and escape from the lattice, giving rise to the serious irreversible release of oxygen. When U ≈ D/2, the d bands in the antibonding (MO)* states overlap with the O 2p non-bonding band, which stimulates the anion/cation redox mechanism and stabilizes the Fermi level via the decrease in local symmetry in the case of electron loss. Therefore, it is necessary to choose appropriate TM-O hybridization, local coordination and a combination to improve the possibility of reversible anion redox in the actual electrochemical window.
Jahn-Teller effect
The LiMn6 superstructure formed by Mn plays an important role in motivating anion redox in LRMO cathode materials. Simultaneously, the anions in turn activate Mn to participate more in the redox reaction. The coupling of Mn and oxygen is supposed to have significant synergistic effects on stabilizing (O2)n- species and activating more Mn ions to participate in the electrochemical process. However, the valence of Mn undergoes a deep evolution in a wide voltage window of 2.0-4.8 V. The layer structure of LRMOs gradually transforms into the Li2MnO4 spinel phase with cycling, which induces Jahn-Teller distortion[33]
The Jahn-Teller effect refers to the asymmetric occupation of electrons in degenerate orbitals that causes a distortion of the geometric configuration, thus reducing the symmetry of the molecule and the orbital degeneracy, and further decreasing the energy of the system. In a Mn3+O6 octahedron, there is only one electron in the doubly degenerate eg orbitals (dx2-y2 and dz2 orbitals) for high-spin Mn3+[34], which leads to asymmetry of the electron distribution. To reduce the energy of the system, the bond length of Mn-O along the z-axis is elongated and four Mn-O bonds in the horizontal direction are shortened, causing a structural distortion that is not observed in the Mn4+O6 octahedron. It is generally considered that the critical mean valence of Mn is 3.5+ when Jahn-Teller distortion occurs. Moreover, unstable Mn3+ is prone to undergo a disproportionation reaction of Mn3+→Mn4+ + Mn2+. Mn2+ dissolves into the electrolyte and deposits on the anode, resulting in a loss of active material and an increase in the electrochemical impedance on the electrode interface. Therefore, it is necessary to regulate the variation of Mn valence during the electrochemical process to decrease the content of Mn3+.
Local structure
Crystal structure and nanodomains
Owing to the composition of the R$$\bar 3$$m LiMO2 phase and C2/m Li2MnO3 component, LRMO materials display complex structural characteristics. The (001) crystal plane of the C2/m phase just overlaps with the (003) crystal plane of the R$$\bar 3$$m phase and both the interplanar spacings are close to 4.7 Å, which causes the two components to appear dissolved at the atomic scale and mixed at the nanometer scale [Figure 2A]. Interestingly, weak peaks at 20-25° appear in the X-ray diffraction (XRD) patterns, owing to the loss of the systematic absence for the LiMn6 superstructure. Jarvis et al.[35] demonstrated that Li[Li0.2Ni0.2Mn0.6]O2 was composed of a solid solution with C2/m monoclinic symmetry and multiplanar defects via the combination of atomic-scale high-angle annular dark-field aberration-corrected scanning transmission electron microscopy (HAADF-STEM) and XRD. In particular, there are two-phase regions, owing to the loss of ordering between Li+ and Mn4+ in the TM layer with less lithium. Yu et al.[36] also clarified the coexistence of LiMO2 and Li2MnO3 with sizes of 2-4 nm in Li1.2Ni0.15Co0.1Mn0.55O2. Yu et al.[37] revealed the symbiotic heterogeneous interface of rhombohedral LiMO2/monoclinic Li2MnO3 at the atomic scale in
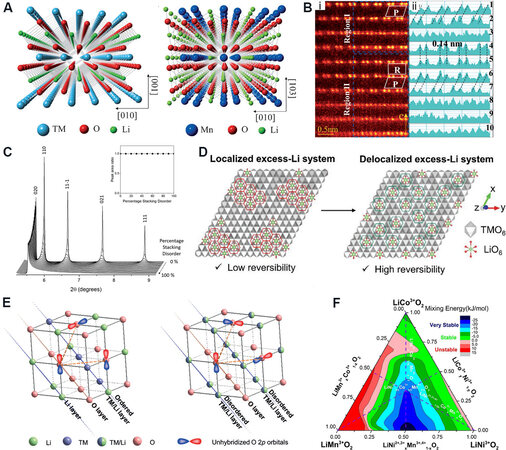
Figure 2. (A) Crystal structure of rhombohedral LiMO2 (left) and monoclinic Li2MnO3 (right)[37]. (B) HAADF images (i) and intensity profiles (ii) of the two-phase intergrowth and heterointerface along the [001]rh zone axis direction[37]. (C) Increasing stacking disorder results in asymmetric peak broadening with no decrease in the peak area ratio between the superstructure peaks and the 001 direction (inset)[41]. (D) Morphological and structural designs for a delocalized excess Li system for enabling high electrode density and reversibility[20]. (E) 2D-ordered cation layered structure with coplanar unhybridized O 2p orbitals (left) and 3D-disordered cation framework with a random spatial distribution of unhybridized O 2p orbitals (right)[49]. (F) Mixing energy predicted from bonding model for the solid-solution LiMO2 (M = Ni, Co and Mn) phase[51]. HAADF: High-angle annular dark-field.
In addition, stacking faults[38,39] along the c-axis exist both in the two-phase symbiotic heterogeneous interface and simple structure, which is the root cause of broadened superstructure diffraction peaks. Shunmugasundaram et al.[40] discovered that the location of the superstructure diffraction peaks varied monotonously with the Ni content, while the probability of stacking faults increased monotonously with the content of Ni according to the XRD patterns fitted by FAULTS. Therefore, the presence of heterovalent ions, such as Ni2+, disturbs the stacking order along the c-axis and broadens the superstructure peaks of the material [Figure 2C]. Similarly, House et al.[41] determined 20% stacking faults in a pristine
Cation arrangement
The LiMn6 superstructure is the key factor to induce anion redox and O-O dimers in the TM layer and its distribution deeply affects the whole structural evolution and electrochemical performance of the material[43]. Hwang et al.[20] discovered that Li1.11Mn0.49Ni0.29Co0.11O2 with a relatively delocalized distribution of the superstructure exhibited more stable lattice oxygen [Figure 2D], which was attributed to the more dispersed distribution of highly active oxygen in an oxidized state that effectively suppressed the polymerization of O-O dimers and the release of O2 during the extraction of Li+. In addition, Song et al.[44] found that a disordered distribution of Li/Mn in the TM layer improved the symmetry of LRMOs and substantially adjusted the redox activity of oxygen, which helped to realize the reversibility of the structure, thereby alleviating the Jahn-Teller effect, decreasing the band gap and expanding the two-dimensional (2D) diffusion channel of Li+. The Li vacancy in the superstructure inhibited the dimerization of O-O in anion redox. This disordered arrangement is also universally applicable to other Li-rich systems. For Li2RuO3, the local symmetry around the oxygen ions is adjusted by the disordered arrangement in the Li/TM layer, so that the response structure to anion redox of the material transforms from the original O-O dimers to the retractable O-Ru-O configurations[45], thereby inhibiting the significant release of oxygen and promoting the cycle stability.
The cation arrangement between the TM and Li layers profoundly influences the coordination environment of oxygen and the whole structural stability of the material[46-48]. In LRMO materials with 2D cation arrays, the oxygen ions in Li-O-Li configurations interact with adjacent oxygen ions to reduce the O-O distance and their energy in the oxidized state. The interaction of different oxygen ions leads to a decrease in the bond angle of O-TM-O and the overlap of TM eg and O 2p orbitals[49], which weakens the bonding of TM-O and damages the stability of the structural framework. Two unhybridized O 2p orbitals are non-coplanar in the structure with disordered three-dimensional (3D) cation arrays and the lack of interaction and bonding between the two orbitals inhibits the dimerization of O-O. This results in a relatively stable lattice structure of oxygen [Figure 2E] that hinders the diffusion and migration paths of Li+. Liu et al.[47] synthesized Ti-doped Li1.2Ti0.26Ni0.18Co0.18Mn0.18O2, where Li+ was partially replaced by Ti4+, and the Li/Ti cation disorder structure strengthened the tolerance of deformation and restricted the migration of TM ions in the TM layer during the delithiation process. This contributed to lowering the electrochemical reaction potential and improving the reversibility of anion redox. Lee et al.[50] certified that the structural stability of the bulk and surface was enhanced with an increasing degree of cation disorder in Li1.2Ni0.2Mn0.6O2 during the initial activation process. This ultimately inhibited the structural deterioration with subsequent cycling and maintained the electrochemical activity and rate performance even in the case of cation disorder.
Distribution of elements
The distribution of elements in pristine particles affects the structure and electrochemical properties of LRMOs. Liang et al.[51] proposed a TM bond model and predicted the order of the strength of TM-TM bonds (Mn4+Mn4+ > Ni2+Mn4+ > Ni3+Mn4+ > Co3+Mn4+ > Co2+Mn4+ > Ni2+Ni4+) by studying the interaction and distribution of TM elements [Figure 2F]. It was found that Co and Mn could easily segregate to form clusters in the Ni environment, which was a significant reason for the deterioration of the structure and the degradation of the electrochemical performance during cycling. Adjusting the interaction of TM elements and promoting the uniformity of elements can prominently promote the structural stability of layered NCM materials. This mechanism is also applicable to LRMO cathode materials, with the segregation of Ni on the surface impeding the migration channel of Li+ and weakening the interaction of Ni-Mn, thereby causing the reduction of manganese ions and the fast degradation of voltage and capacity. In contrast, the uniform distribution of elements at the atomic scale enhances the interaction of TM elements, stabilizes the crystal structure and contributes to excellent capacity retention and more smaller voltage degradation[52,53].
For LRMOs, the distribution of elements can be locally adjusted to produce outstanding electrochemical performance[54,55]. The gradient structure that Mn gradually decreases and Ni and Co gradually increase from the center to the surface with a certain slope inhibits the transformation to the spinel phase and the Jahn-Teller effect, thereby markedly reducing the degradation of voltage, which promotes the cycle performance. In Ni-rich layered materials, reducing the content of Ni in the surface layer and increasing the concentration of Mn[56] not only maintains the high capacity of the center of the material but also improves the stability of the surface layer. This seems to imply that a similar content of TM components can improve the structural tolerance of the material. The inhomogeneous distribution of Li also significantly influences local anion redox. Zhu et al.[57] synthesized an LRMO material with a Li-poor shell, which inhibited the generation of O-O dimers on the surface and the release of O2.
Stoichiometry
C2/m:R$$\bar 3$$m
The Li2MnO3 phase with the C2/m space group as the sole component of an LRMO endows the material with the “Li-excess” characteristic and profoundly affects its structure and electrochemical performance[9]. Li is extracted from the lattice in the form of “Li2O” and leaves the MnO2 phase to stabilize the structure, enabling the composite LRMO layered material to maintain good tolerance to high voltage. Yang and Xia[58] doped NCM811 cathode materials with a certain Li2MnO3 component to sufficiently inhibit the phase transition between the H2 and H3 hexagonal phases with distinctly different cell parameters, which dramatically decreased the variation of the structure volume and alleviated the drastic structural deterioration and capacity degradation. More importantly, the non-activated Li2MnO3 can store the excess lithium, so that Li+ in the TM layer migrates to the adjacent Li-deficient layer during charging to stabilize the structure and realize the sufficient release of carriers and capacity.
The pristine layered structure of Li2MnO3 transforms to the spinel phase during cycling and the capacity source changes to maintain the capacity. However, the difference in the voltage plateau between the two phases leads to voltage degradation, which is a unique phenomenon and problem of LRMOs. The intrinsic atomic structure and arrays of the Li2MnO3 component significantly influence the electrochemical performance. Song et al.[59] found that the structural defects in different orientations and the imperfect atomic arrays of the Li2MnO3 component caused an increase in atomic defects and the concentration of vacancies, ultimately leading to the breaking of grains and performance degradation. The sluggish electrochemical kinetics of the Li2MnO3 phase restrict the rate performance and also limit the performance release of the R$$\bar 3$$m component. However, the control of the current density can regulate the activation of
The introduction of the R$$\bar 3$$m component into Li2MnO3 (via the doping of Ni or Co) can significantly improve the irreversible oxygen loss. It has been shown that the intrinsic charge compensation of anions for Li2MnO3 leads to the completely irreversible release of O2, but the irreversible oxygen loss accounts for 25% in Li1.2Ni0.13Co0.13Mn0.54O2 with a Li2MnO3 ratio of 50%. This means that Ni and Co play an important role in stabilizing lattice oxygen in the bulk material. Liu et al.[60] studied the electrochemical properties of
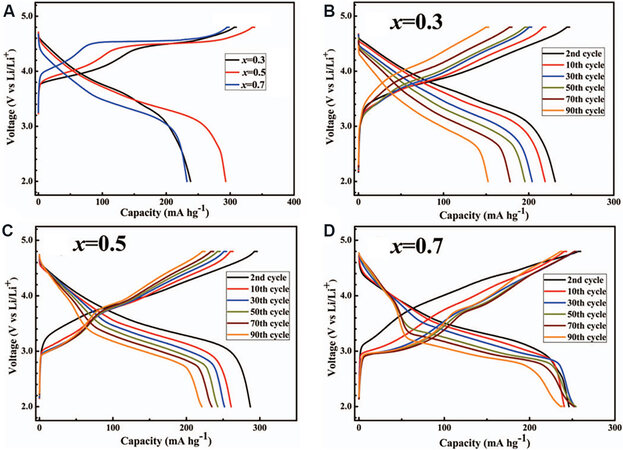
Figure 3. Typical charge-discharge curves of (A) initial and (B-D) cycling process for xLi2MnO3·(1-x)LiNi1/3Co1/3Mn1/3O2 (x = 0.3, 0.5 or 0.7) cathode materials[60].
Ratio of Ni, Co, Mn and Li/O
Every element and stoichiometric ratio play a critical role in determining the whole structure and electrochemical activity[62]. Hua et al.[63] systematically studied the relationship between Li content and the structure of LixNi0.2Mn0.6Oy by matching the Li content with the precursor. When x < 0.4, the material was indexed to the spinel phase (Fd$$\bar 3$$m), while at 0.4 < x < 1.2, the material was a complex of the spinel (Fd$$\bar 3$$m) and rock salt (Fm$$\bar 3$$m) phases, including Li and the layered monoclinic phase (C2/m). At 1.20 < x < 1.52, the material was indexed to the pure monoclinic phase (C2/m). The ratio of Li/O represents the content of Li-O-Li configurations in the LRMO material, thereby affecting the anion redox and the structure of the energy bands[64]. The lower ratio of Li/O means the TM-O hybridization and non-bonding O 2p bands move to lower energy levels, which contributes to a higher potential of redox reaction that promotes the electrode reaction kinetics, reversibility and energy efficiency [Figure 4A]. The higher ratio of Li/O accelerates the anion redox reaction, inevitably introducing an excess disordered structure that is regarded as the buffer regulating the conflict between oxygen evolution and the crystal structure and making the LRMO more stable and flexible[65].
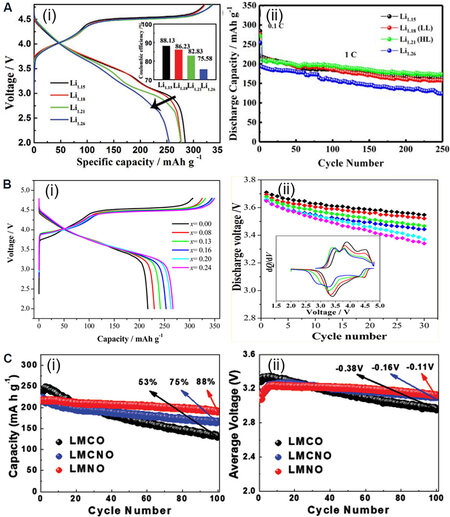
Figure 4. (A) Initial charge-discharge plots with Coulombic efficiency (i) and cycle performance (ii) of the as-prepared
The regulation of lithium and oxygen is also applied to other cathode materials. Xiao et al.[38] designed different cooling technologies and synthesized Li2MnO3 with various nonstoichiometric oxygen defects and stacking faults. These defects decreased the valence of Mn and the increase in Mn3+ content accelerated the activation of Li2MnO3, but this simultaneously led to a more rapid structure deterioration to the spinel and capacity degradation. Wu et al.[66] introduced an appropriate excess of lithium into NCM811, resulting in the reduction of partial Ni3+ to Ni2+ and the decrease of Li/Ni disorder, which improved the electrochemical stability and structural robustness during cycling. Ji et al.[67] designed a novel cathode material with excellent rate performance and reversible redox of oxygen via the introduction of excess Li into spinel LiMn2O4. The cations over stoichiometric ratio and the derived local disorder eliminated the typical first-order phase transition reaction of the ordered spinel material and realized higher practical capacity.
The oxidation of oxygen in Li2MnO3 during charging generally causes the irreversible loss of lattice oxygen. However, the introduction of Ni and/or Co can dramatically inhibit the loss of oxygen, which results in the formation of Li[Li0.2Ni0.2Mn0.6]O2 and Li[Li0.2Ni0.13Co0.13Mn0.54]O2. The introduced Ni is enriched on the particle surface to form Li-deficient and Ni-rich rock salt shells as protection layers to inhibit oxygen loss[68]. Though the addition of Co cannot cause the formation of the rock salt phase, it still restricts the oxygen loss overall. In addition, the overlap of the Co3+/4+ t2g and O 2p bands and the intense covalency of Co-O in LRMO can promote the coupling between Co and O and the anion redox[69]. Nevertheless, this leads to an increase in Mn3+ content, a decrease in electrochemical reaction potential, the generation of oxygen vacancies and the migration of TM ions to the Li layer[70], which accelerates the capacity and voltage degradation [Figure 4B]. Accordingly, the appropriate increase in Ni content and a lower ratio of Co/Ni can efficiently adjust the coordination environment and local electron structure of oxygen to reduce the proportion of Mn3+/Mn4+ redox couples at low voltages and enhance the operation voltage[19]. This also enables the TM-O and non-bonding O 2p bands to move down to lower energy levels, thereby inhibiting anion redox and the corresponding phase transition and efficiently improving the structural stability and capacity/voltage retention. Enhancing the Ni content not only changes the center of redox in LRMO cathode materials but also promotes the reversibility of Mn migration[71] by a shielding effect, where the random transition of Mn is restricted in a certain region by the electrostatic shielding effect of Ni ions [Figure 4C].
Particle morphology
The particle morphology of a cathode material plays an important role in the transportation of Li+, the release of capacity, the stability of the structure and the stress-strain relationship. At present, the mainstream particle morphology of layered cathode materials includes irregular single-crystal particles (nano- and microscales), secondary spherical particles consisting of primary single-crystal particles and other single-crystal or polycrystalline particles with special morphologies. Submicron single-crystal particles possess high specific surface area and shorter Li+ diffusion path, which enables sufficient contact with the electrolyte and inhibits the generation of local nanodomains inside the particles. These advantages contribute to the sufficient release of electrochemical activity to ultimately realize excellent reversible capacity and rate performance. However, the severe release of O2 on the surface and the side reaction with the electrolyte lead to the formation of a cathode-electrolyte interface (CEI) that impedes the transportation of Li+. The oxygen vacancies on the surface cause destruction of the lattice and densification of the structure, giving rise to a serious solution of TM ions that further deteriorates the capacity retention and rate performance. With cycling, the smaller particles also undergo severe structural deterioration and performance degradation. Smaller sized particles cannot meet the requirements of tap density in industry. Microscale single-crystal particles efficiently alleviate the side reaction with the electrolyte at the interface via a decrease in the specific surface area, thereby maintaining the structural integrity of the inner component. However, the poorer Li+ mobility limits the release of capacity and the utilization of the material, which may induce the generation of local domain[72,73] boundaries that hinder the transportation of Li+, leading to structural deterioration. Thus, it is necessary to balance the particle size and the electrochemical performance.
In typical layered cathode materials, the diffusion path of Li+ is the 2D channel vertical to the c-axis in the Li layer, i.e., the migration direction of Li+ is vertical to {010} crystal plane, including the (010), ($$\bar 1$$10), ($$\bar 1$$00), (0$$\bar 1$$0), (1$$\bar 1$$0) and (100) crystal planes. However, the close packing of TM and O in the (001) planes severely hinders the transportation of Li+. Therefore, the exposed planes in the particles are key to determining the extraction/insertion rate of Li+[74]. In order to obtain excellent Li+ migration rates, the {010} crystal planes should be more exposed and the proportion of (001) planes reduced[75] [Figure 5A]. However, the thermodynamic surface energy of the close-packed (001) plane is the lowest and generally the proportion of the exposed (001) plane is the largest, which creates difficulties in providing fast transportation channels and planes for Li+. To enhance the exposure rate of {010} crystal planes, it is necessary to reduce the surface energy of {010} and increase the surface energy of the (001) plane during the nucleation and growth process. The exposure of special planes enables the particles to exhibit special morphologies, such as nanosheet-/plate-like single crystals and their stacked spherical[18] [Figure 5B], 3D orthogonal and cubic maze-like polycrystals[76].
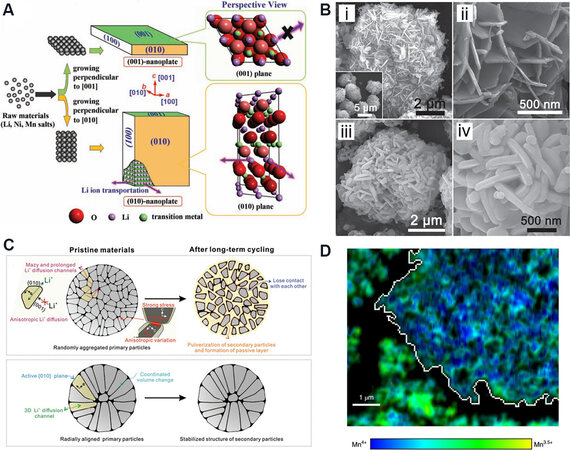
Figure 5. (A) Schematic illustration of two kinds of nanoplates and the microstructure of their surfaces[75]. (B) Field emission scanning electron microscopy images of (i) precursors and (ii) their magnified image, (iii) hierarchical structured lithium-rich material after pre-heating at 500 °C and (iv) its magnified image[18]. (C) Schematic illustration of the structure and characteristics of commercial NCM (top) and the materials with radially aligned single-crystal primary particles (bottom)[79]. (D) A single large agglomerate (to the right of the image, outlined in white) that is substantially more oxidized than the surrounding areas[80].
Currently, secondary spherical particles consisting of primary single-crystal particles are widely applied and their advantages include the side reaction at the interface, owing to the low specific surface area, and the better rate performance than that of single-crystal particles of the same size. In addition, the stacking of particles with different sizes leaves the spatial occupation denser, thereby exhibiting a higher tap density. The connection between the primary particles is also worthy of consideration. Appropriate pores are conducive to the sufficient contact between the electrode particles and the electrolyte. It can also ensure the effective connection between particles, so that electrons and ions can be facilely transferred in solid-phase materials[77,78]. However, the anisotropic volume changes of the primary single-crystal particles during charging cause stress/strain and cracks in the whole polycrystalline particles, leading to an obstruction in Li+ diffusion and an increase in impedance. Ultimately, the particles become broken, resulting in the dissociation of active materials in the electrolyte and dramatic performance deterioration. The anisotropic volume change can be improved by the morphological control of the primary particles.
Xu et al.[79] synthesized radially-oriented single-crystal primary nanosheets for Ni-rich cathode materials via the optimization of synthesis technology [Figure 5C]. The exposed {010} planes and the morphology extending from the surface to the center ensure the rapid diffusion of Li+. The radially-oriented primary particles with consistent crystal orientation can regulate the volume change during cycling to alleviate the stress/strain, contributing to excellent structural stability and cycle performance. Furthermore, the permeation of the liquid electrolyte in broken particles results in a larger contact area between the active material and electrolyte, decreasing the charge transfer resistance and enhancing the apparent diffusion coefficient of Li+, eventually realizing high reversible capacity and outstanding rate performance. This special primary particle morphology is also worthy of study and applying to the morphological design of LRMO materials. In addition, Csernica et al.[80] analyzed the oxygen evolution in cross sections of LRMO particles with different sizes in both the inner section and on the surface. It was found that the degree of oxygen evolution of the single primary particles was highly dependent on their place in the secondary structure, i.e., the closer the primary particle approached to the surface, the severer the oxygen evolution and structural transformation [Figure 5D]. Primary particles may be important factors in determining the overall capacity of a material compared with secondary particles from a morphological perspective. It was supposed to explore much larger secondary LRMO material particles to relieve the release of oxygen.
TEMPORAL DIMENSION
In this section, we focus on the charge compensation, element migration, phase transformation and reconstruction of the energy band structures of LRMO materials during the charge and discharge process.
Initial charge and discharge
Charge compensation mechanism
The LRMO cathode material xLi2MnO3·(1-x)LiMO2 possesses a superior discharge capacity of over
According to the absorption edge analysis of each element at different states of charge [Figure 6A], it is found that the slope below 4.4 V during charging corresponds to the charge compensation of Ni2+/Ni4+ and Co3+/Co4+ without the variation of the Mn4+ valence, which is attributed to the charge compensation mechanism of the LiMO2 component[9]. The absorption edge of the TM at 4.5 V shows that there is no change in the K edge of Ni and Co[81]. The Mn K-edge X-ray absorption near-edge structure spectrum indicates that the environment of Mn changes at 4.5 V according to the continuous variation of the absorption edge, which results from the distortion of MnO6 octahedra. Hence, the lattice oxygen coordinated with Mn can more easily participate in charge compensation and generate electron holes. The unique anion charge compensation of LRMOs occurs in the plateau region at 4.5 V (the triggering condition has been discussed above), which is ascribed to the Li-O-Li configurations and the hybridization conditions of TM-O.
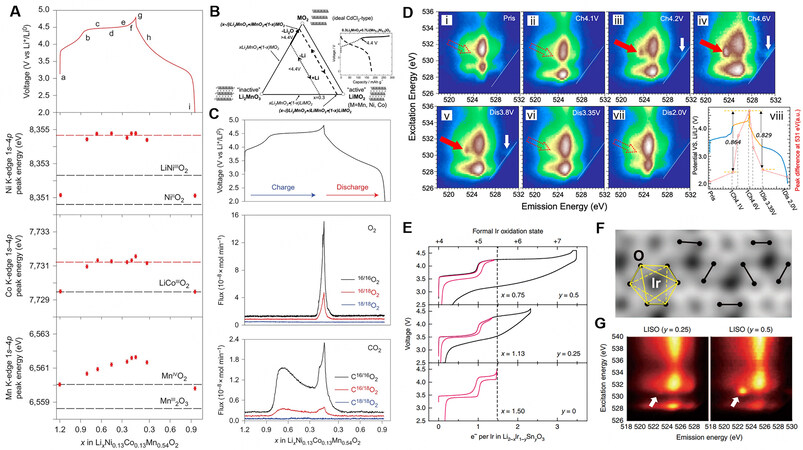
Figure 6. (A) 1s-4p peak energy in the initial cycle showing almost no change in the environment around Ni and Co on charging across the 4.5 V plateau[83]. (B) Compositional phase diagram showing the electrochemical reaction pathways for a xLi2MnO3·(1-x)LiMO2 electrode[9]. (C) Operando mass spectrometry of 18O-labelled Li1.2Ni0.13Co0.13Mn0.54O2 cathode during the initial cycle[83]. (D) O K-edge mRIXS of a series of Li2RuO3 electrodes at different electrochemical states. The solid red arrows indicate the striking, sharp features of oxidization at the high voltage state. (i-vii) represent the mRIXS of Li2RuO3 at the state of the pristine, charged to 4.1 V, charged to
In the initial cycle, oxygen evolution occurs in the Li2MnO3 component with the extraction of Li+ manifesting as the first-order phase transition reaction in the form of “Li2O” net loss [Figure 6B], which involves the nanodomains and dislocations formed by localized uneven delithiation[82]. It is clarified that CO2 gas forms at the beginning of the voltage plateau by operando mass spectrometry and oxygen isotope labeling[83], which can be attributed to the oxidic lattice oxygen with high activity reacting with the electrolyte (possibly including the decomposition of residual Li2CO3 on the surface). Subsequently, O2 escapes from the lattice at the end of the voltage plateau[84], which does not derive from the oxidation of the electrolyte [Figure 6C]. However, the amount of oxygen release is considerably lower than the estimated value generated by the activation of the Li2MnO3 component. Considering that O2 cannot migrate from the bulk to the surface, the emission of O2 occurs only at the near surface. According to calculations, the equivalent thickness of the surface where oxygen is released is ~2-3 nm[85].
The anion charge compensation mechanism is distinct for different lithium-rich materials[28]. For Li2RuO3, the cation Ru4+/Ru5+ and anion O2-/O2n- couples reversibly participate in charge compensation [Figure 6D], owing to the d-sp hybridization related to the redox coupling without irreversible oxygen evolution. For
Energy band reconstruction
The inconsistency of the initial charge/discharge curves certainly means the reconstruction of the bulk structure and the corresponding energy band. According to the above discussion, the redox of Ni and Co is the same as the charge compensation of LiMO2 with good reversibility, which cannot influence the structure and energy band. Therefore, it is the key to defining the anion charge compensation mechanism. It is found that the valence of Ni and Co remains unchanged at the beginning and the end of discharge and the variation of valence during discharging is reversible via the comparison of the absorption edge for Ni, Co and Mn. However, the valence of Mn decreases slightly, which may be ascribed to the activation of Mn in the Li2MnO3 component[86]. Based on the test and calculation results, more than half the discharge capacity (~280 mAh g-1) is still unidentified. If we exclude the side reaction with the electrolyte, this part of the capacity can be regarded as the contribution from the reduction of oxygen.
In order to verify the reversibility of anion redox, Li et al.[87] determined the reversible evolution of O-O dimers during charging for the first time via in-situ surface enhanced Raman spectroscopy. The results seemed to suggest the reversibility of the redox behavior of oxygen but failed to explain why the reduction plateau of oxygen disappeared during discharging. Yabuuchi et al.[84] considered that oxygen was electrochemically reduced on the electrode surface below 3 V during discharging. Sun et al.[64] demonstrated that there was larger voltage hysteresis (≈ 0.5 V) during the anion redox via adjusting the cut-off voltage. Gent et al.[88] revealed that the strong couple effect between anion redox and the partially reversible cation migration contributed to the decrease over 1 V in the reduction potential of the bulk oxygen, causing the rearrangement of the anion and cation redox potentials.
However, this does not seem to explain intuitively and thoroughly the root cause of the energy band structure rearrangement. Considering the irreversible release of O2 in the near-surface region of the lithium-rich material during the first charging process, it should be questioned whether O2- will also dimerize to form O2 molecules after being oxidized inside the particle body, which are restricted in the crystal lattice and cannot escape. This assumption seems more reasonable. Recently, House et al.[41] used high-resolution RIXS and 17O magic angle spinning nuclear magnetic resonance (NMR) spectroscopy to successfully confirm the existence of O2 molecules in the crystal lattice instead of O22-, longer peroxide-like O-O bonds or electron holes on O2-. In addition, they calculated and simulated the intralayer migration and disordered arrangement of TM elements in the fully charged state, which destroyed the LiMn6 honeycomb ordered superstructure and formed vacant clusters that can accommodate O2 [Figure 7]. The migration in the TM layer is irreversible. During the discharge process, the coordination environment of active oxygen changes from O-Li4Mn2 to O-Li6 with the insertion of Li+, which increases the number of oxygen non-bonded orbitals and increases the energy level[14]. Therefore, the reduction of oxygen occurs in the low voltage stage, which leads to a larger voltage hysteresis. The above perspective is currently the most satisfactory conclusion of the reconstruction of the structure and the rearrangement of the energy band during the first cycle of LRMO materials, but it still requires the unremitting efforts of researchers to verify and improve it.
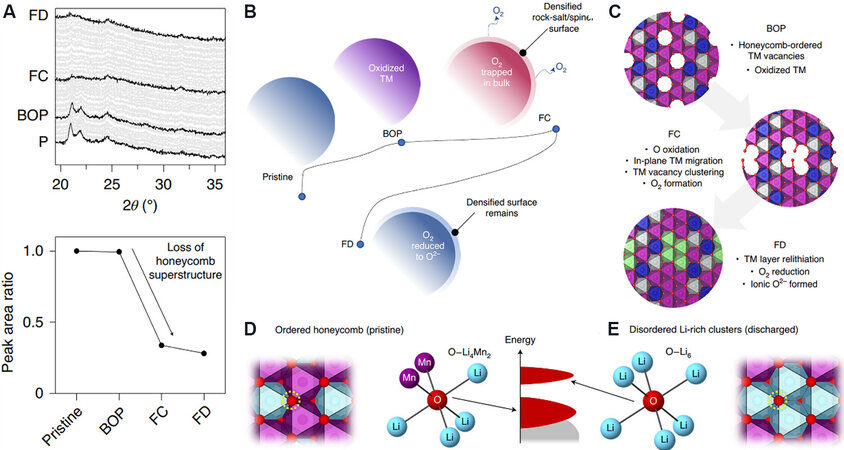
Figure 7. (A) Operando XRD of Li1.2Ni0.13Co0.13Mn0.54O2 at different stages of the initial cycle, where P is pristine, BOP is the beginning of the plateau, FC is full charge and FD is full discharge. The peak area ratio of the superstructure peaks relative to the 001 peak decreases across the plateau, which indicates the loss of the honeycomb structure[41]. (B) Macroscale changes of the cathode particles during the first cycle[41]. (C) Structures of atomic-scale ordering changes within the TM layer modeled by DFT[41]. (D) Local coordination environment around oxygen in pristine honeycomb-ordered O-Li4Mn2[14]. (E) Local coordination around oxygen in disordered Li-rich cluster O-Li6 after the first cycle. O 2p states belonging to O and that have six coordinating Li are higher in energy than those that have four coordinating Li, which results in the initial voltage hysteresis[14]. XRD: X-ray diffraction.
Element migration
The topological reaction occurs before 4.4 V, where Li+ ions are extracted from the LiMO2 component with no variation in the frameworks[89]. During the electrochemical process, the Li+ ions in the LiMO2 component are gradually consumed, so the Li+ ions in Li2MnO3 migrate from octahedral sites in the Mn layer to tetrahedral sites in the Li layer to participate in charge compensation, which provides extra binding energy to stabilize the structure. Li+ ions continue to be extracted from the Li2MnO3 component with a series of complicated element migrations and phase transition processes at the 4.5 V voltage plateau[90,91]. The charge compensation of oxygen ions at the 4.5 V plateau leads to TMO6 octahedral distortion[92] and even the formation of oxygen vacancies. Taking the Li1.5MnO3 model containing Li vacancies, Yu et al.[93] found that the migration energy of Mn from the 4g site to the 2b Li site was ~3.07 eV through theoretical calculations. With the introduction of oxygen vacancies, the migration energy was rapidly reduced to 1.86 eV, which promotes the migration of Mn ions.
It is generally considered that partial Mn ions migrate from octahedral sites to the coplanar tetrahedral sites in the Li layer and ultimately from tetrahedral sites to Li sites at this stage. Radin et al.[94] claimed that the Mn migration to tetrahedral sites was actually indexed to the oxidation of Mn4+/Mn7+ simultaneously triggering the generation of O2 molecules and peroxide ions [Figure 8A] according to first-principles calculations based on the Li-Mn-O phase diagram. Mn7+ was then reduced and returned to the original octahedral sites and there were also Mn7+ ions in tetrahedral sites at the end of charging. Kleiner et al.[95] quantified the migration track of TMs via synchrotron radiation XRD differential Fourier fitting. It was found that ~8% TMs occupied the tetrahedral sites in the Li layer during charging and ~2%-5% TMs occupied Li octahedral sites without the occupation of tetrahedral sites after discharge. Their results strongly suggested that 3%-6% TMs can reversibly migrate from the tetrahedral sites to the original TM octahedral sites during delithiation [Figure 8B].
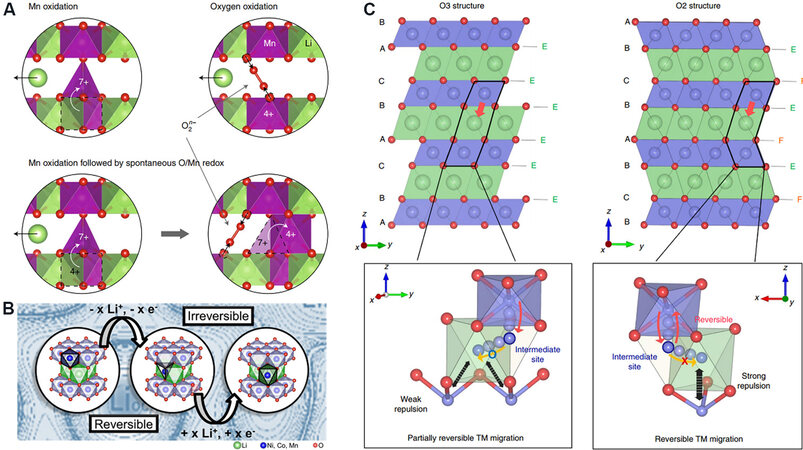
Figure 8. (A) Alternative charge mechanisms in LRMO cathode materials. Oxidation of Mn4+ to Mn7+, followed by the spontaneous formation of peroxide or trapped oxygen molecules accompanied by the reduction of Mn[94]. (B) TM migration in an LRMO from the octahedral TM sites via tetrahedral sites into octahedral Li sites[95]. (C) Comparison of crystal structures. Schematic illustrations of crystal structures of O3-type (left) and O2-type (right) LRMOs with TM migration paths. Although the TM ions in the O3 structure can readily occupy Li sites that share only edges with neighboring cations, the TM ions in the O2 structure are subject to strong repulsion when they occupy Li sites that face share with neighboring cations[98]. LRMOs: Lithium-rich manganese-based layered oxides.
The TM ions partially migrate to Li sites, leading to the formation of localized spinel-like phases. Therefore, the electrochemical activity of Mn is activated and Mn will be excessively reduced to Mn3+ below 3 V. This tends to stimulate the Jahn-Teller effect, as well as the disproportionation of Mn3+, resulting in structural deterioration and element dissolution. In particular, in the near-surface layer of LRMOs, TM migration and structural reorganization are more serious due to the O2 release and the destruction of the oxygen framework, resulting in the formation of disordered rock-salt phases and surface densification[91]. Therefore, irreversible element migration is a vital reason for the phase transition and the degradation of electrochemical performance. It is particularly important to suppress the irreversible migration of TMs, especially the migration process from the transition-state tetrahedral site to the Li octahedral site. The O2-type cathode material[96,97] then emerges and the special oxygen stack (ABCBA) enables TM ions to undergo intense electrostatic repulsion when occupying the octahedral Li sites coplanar with adjacent TM cations. This effectively suppresses the irreversible TM migration and phase transition[98] [Figure 8C]. Furthermore, if the TMs can realize reversible migration between the TM and Li layers during the charge-discharge process, it is also worthy of affirmation. Ma et al.[99] clarified that Mo can reversibly migrate between the Li and TM layers in Li2MoO3, with the material maintaining good structural and electrochemical stability with the “unit cell breathing”[12] mechanism.
Electrochemical cycling
Although the anion redox mechanism in LRMO cathode materials provides considerable specific capacity, its instability and irreversible damage to the structure have a profound impact on the cycle performance of the materials and cause serious problems, including capacity attenuation, voltage decay and hysteresis. The following sections describe the key issues during cycling from different perspectives.
Oxygen vacancy transfer and element segregation
As LRMOs are cycled under a wide voltage range of 2-4.8 V and high voltage conditions, the lattice oxygen redox and complex structural evolution can continue to exist. From the above discussion, the TMs in the TM layer not only migrate to the Li layer after the first activation but will also migrate in the layer to form localized nanopores to restrain O2 molecules. Moreover, the oxygen ions around the nanopores become more active due to the change in the coordination environment and the increase in the number of non-bonded 2p orbitals, which in turn makes it easier to form oxygen vacancies and promote the migration of TMs[100]. The nanopores gradually expand and merge [Figure 9A], and around the pores, the continuous transfer of TMs leads to segregation and densification of the structure. This phenomenon continues with charge and discharge cycling. In this case, the active lattice oxygen gradually forms O2 molecules and gathers in large pores, which cannot provide capacity. The densified rock salt phase structure around the pores is also electrochemically inert, leading to continuous capacity attenuation, which also hinders Li+ transport and charge transfer and increases electrochemical impedance and voltage hysteresis.
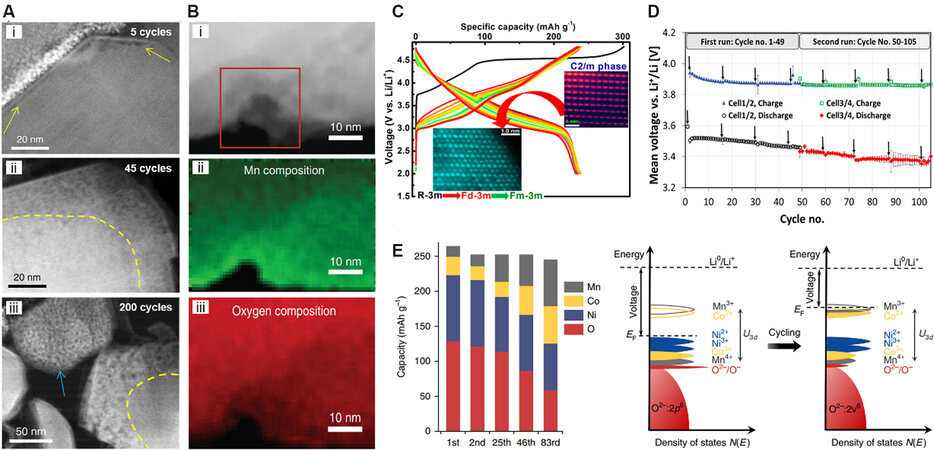
Figure 9. (A) HAADF-STEM images of Li1.2Mn0.6Ni0.2O2 cathode showing the gradual propagation of the nanovoid-populated zone from the particle surface towards the interior of the particle with increasing cycles[101]. (i-iii) Represent the HAADF-STEM images of the material after different cycles (5 cycles, 45 cycles, 200 cycles, respectively). (B) Annular dark-field STEM image (i) of an opened pore. Mn (ii) and oxygen (iii) composition extracted from the quantification of the electron energy-loss spectroscopy map[15]. (C) Charge-discharge curves and phase structural evolution of LRMO during electrochemical cycling[105]. (D) Mean discharge/charge voltage of the two cells cycled simultaneously (cell 1/2: 22 °C, cycles 1-49; cell 3/4: 25 °C, cycles 1-49; 22 °C, cycles 49-105), with the error bars representing the standard deviation between the two cells[95]. (E) Contribution towards the discharge capacity from each element at various cycles (left). An illustration of the Fermi level being lifted up as a result of the electronic structure change (right)[15]. HAADF-STEM: High-angle annular dark-field aberration-corrected scanning transmission electron microscopy.
In addition, there are differences in oxygen release on the surface and inside of LRMO particles.
Local phase structural transformation
During the cycling of LRMO materials, the layered structure gradually becomes unstable with the generation of oxygen vacancies and the migration of TMs. When the TM, especially Mn, occupies the octahedral site of the Li layer, the surrounding re-embedded sites of Li+ are transferred to the tetrahedral vacancy, forming a spinel-like LiMn2O4 phase of a sub-nanodomain. Although a considerable amount of Mn will reversibly return to the original position after it migrates from its own octahedral position to the tetrahedral position of the Li layer in each charge and discharge process and the recurring spinel nanodomain proposed by Xiao et al.[104] appears, there is still some Mn irreversibly migrated to the Li layer. This process continues to accumulate, from the first charge and discharge Li site octahedra are occupied by TMs (mainly Mn) at ~2%, which gradually increases to ~5% after 100 cycles[95]. In the wide voltage range of 2-4.8 V, the Mn ions undergo a profound valence state change. When discharged to ~2.8 V, Li+ ions are excessively embedded in the 16c octahedral position of the cubic spinel LiMn2O4 phase, forming a tetragonal spinel Li2Mn2O4 phase, and the valence state of Mn decreases from +3.5 to +3, resulting in Jahn-Teller distortion and the dissolution of Mn2+ caused by the disproportionation reaction of Mn3+. This leads to further deterioration of the material structure and eventually an inactive disordered rock salt structure (Fm$$\bar 3$$m space group). On the surface of the material, due to the loss of Mn and the migration of Ni, a large number of maligned NiO rock salt phases will be formed[105] [Figure 9C].
The series of phase structural transformations mentioned above lead to a decrease in electrochemically active materials that can accommodate the reversible deintercalation of Li+. In addition, the local disordered rock-salt phase of cations, especially on the surface, seriously hinders the migration of Li+ and charge transfer and increases the electrochemical impedance and voltage hysteresis. Although the production of the active spinel phase during cycling compensates to a certain extent for the capacity decline caused by the gradually weakened anion charge compensation, this leads to another unfavorable factor where the
Electrochemical reaction kinetics
Li+ migration
In layered cathode materials, Li+ mainly carries out 2D interlayer migration in the Li layer. There are two migration paths for Li+ from one octahedral position to the next octahedral position[106] [Figure 10A]. The first is that Li+ directly passes through the center of O-O on the common edge of two octahedrons, which is known as oxygen dumbbell hopping (ODH). The second is to migrate to the tetrahedral site coplanar with the two octahedral by relying on the vacancy left by other Li+ diffusion. It then moves to another octahedral position to complete a migration process known as tetrahedral site hopping (TSH). The diffusion barrier of the TSH path is much lower than that of the ODH path through theoretical calculations. Therefore, when the material is charged in the early stage, Li+ migrates in the ODH path first because there are not many vacancies. With the continuous emergence of Li+ and the increase of vacancies, the migration path of Li+ gradually changes from ODH to TSH and the migration rate is gradually accelerated. Liu et al.[107] pre-introduced Li vacancies in Li-rich materials, which not only reduced the energy barrier of Li+ in the initial ODH diffusion path but also accelerated the transition from difficult ODH to facile TSH.
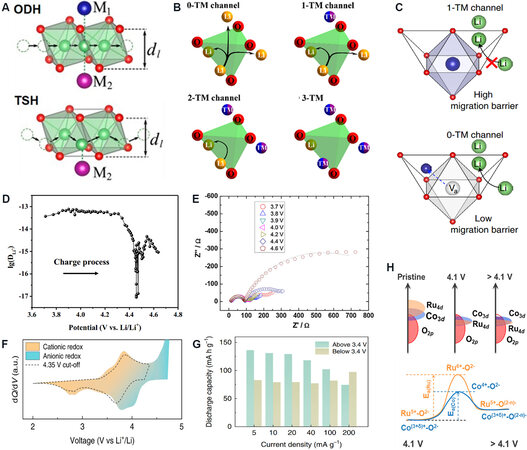
Figure 10. (A) Illustration of ODH and TSH types of Li+ diffusion pathways[106]. (B) Schematic diagram of Li+ distributions around tetrahedral sites in the cation-disordered oxide: 0-TM; 1-TM; 2-TM; 3-TM[108]. (C) Illustration of a TMO6 octahedron with two face-sharing tetrahedral sites. Migration of the TM from the octahedron transforms a 1-TM channel into a 0-TM channel[109]. (D) DLi+ of a LRMO material under different states during the initial charge[110]. (E) Nyquist plots of Li1.2Mn0.56Ni0.16Co0.08O2 electrodes at various potentials[111]. (F) dQ/dV curve of O2-LRMO measured at a current density of 5 mA g-1, which proves the reaction mechanism of anions at high potential[98]. (G) Variation of discharge capacity as a function of current density is estimated for the two classified voltage ranges of 2.0-3.4 and 3.4-4.8 V[98]. (H) Schematic diagrams of density of states at different states of charge (top) and anionic oxygen oxidation coupled with cationic reduction (bottom), showing the effect of different activation energy in the intermediate states on anionic kinetics in Li1.2Ru0.4Co0.4O2[112]. ODH: Oxygen dumbbell hopping; TSH: tetrahedral site hopping.
When a TM occupies the Li site in the crystal structure of the material (especially after cycling and near the surface of the material), the migration channel of Li+ gradually deteriorates from 0-TM to 3-TM[108] [Figure 10B] and the diffusion path of Li+ will not only be blocked, but also suffer from the electrostatic repulsion of the high-valence TM, which greatly increases the migration energy barrier of Li+. Huang et al.[109] found that Cr can realize the non-topotactic reaction of reversible octahedral to tetrahedral migration in the process of delithiation in lithium-rich disordered rock salt materials. Octahedral TMs are the main obstacle to Li+ diffusion. When Cr migrates to the tetrahedral position, it will correspondingly provide a tetrahedral vacancy for Li+, which improves the migration conditions of Li+ [Figure 10C]. This principle can be used for reference to alleviate the deterioration of Li+ diffusion kinetics caused by the deterioration of the layered oxide structure. In addition to Cr, other electronic configurations, such as d5 and d10, are also conducive to the migration to the tetrahedral position, but the matching between the TM size and tetrahedral pores needs to be considered simultaneously.
Anion reaction kinetics
In the first charging activation process, our previous work[110] found that the diffusion coefficient of Li+ (DLi+) increases slightly at the initial charging stage, which corresponds to the release of Li+ from the LiMO2 component. However, DLi+ drops sharply at the 4.5 V plateau [Figure 10D], which corresponds to the charge compensation process of oxygen in the Li2MnO3 component. This anomalous behavior cannot be explained from the perspective of the Li+ migration channel. Although a small amount of TM migration occurred during the process, which hindered the migration of Li+, the generation of Li vacancies and the expansion of c-axis layer spacing were still favorable for Li+ diffusion. Considering the electrochemical redox reaction with both electron and ion conduction, the above apparent decrease in DLi+ only represents a decrease in the electrochemical reaction rate rather than a decrease in the diffusion capacity of Li+. Therefore, the electron transfer of anions is likely to be a limiting factor.
Nayak et al.[111] used electrochemical impedance spectroscopy (EIS) to test the reaction kinetics of a LRMO at various reaction steps at different potentials and found that the charge transfer resistance increased significantly at voltages of > 4.4 V [Figure 10E], indicating that the electrode reaction kinetics at high potentials were controlled by anionic charge transfer rather than Li+ diffusion. Yu et al.[36] used time-resolved X-ray absorption spectroscopy (XAS) to determine the characteristic reaction kinetics of each element. Compared with Ni and Co, the reaction kinetics of the Mn site before and after the initial activation of Li2MnO3 was much worse. These results suggest that Li2MnO3 (anionic charge transfer) is a key component in limiting the rate capacity of LRMOs. Moreover, the anion reaction kinetics in the O2-type LRMO cathode material are also slower than that of the cations. Eum et al.[98] analyzed the dQ/dV of different potential ranges and found that the charge compensation of the anion in O2-type materials during charge and discharge occurs at a high potential [Figure 10F], which is different from the anion reduction in the O3-type material at a low potential. This creates the conditions for simply examining the reaction kinetics of anions and cations by controlling the applied current and analyzing the capacity evolution at different potential ranges [Figure 10G]. In order to improve the reaction kinetics of the anions, Li et al.[112] found that the strong covalent property of Co and O enables the charge compensation of oxygen to occur in advance before the complete oxidation of Co3+ to Co4+. This strong coupling effect makes the rapid redox of Co(3+δ)+/Co4+ a medium to improve the kinetics of anion redox in lithium-rich materials [Figure 10H]. Other anions, such as S2-, exhibit faster reaction kinetics and lower voltage hysteresis than O2- but also lower the redox potential[113] and affect the energy density due to the weaker electronegativity.
PREPARATION AND MODIFICATION
Preparation method
Co-precipitation method
Generally, various metal salts are prepared into a mixed solution according to the target stoichiometric ratio and the metal salt solution and the precipitating agent (e.g., OH-, CO32-, C2O42- and so on) are added dropwise to deionized water simultaneously. An appropriate amount of chelating agent (e.g., ammonia) is added to coordinate with metal ions to buffer the precipitation reaction. Meanwhile, the pH and stirring speed are controlled at a certain temperature to allow metal cations to co-precipitate. The precursor and lithium source (e.g., LiOH, Li2CO3 and so on) are then uniformly ground and then calcined at a high temperature to obtain the final lithium-rich oxide. This method can realize ion mixing on the nanometer scale and the subsequent formation of the phase structure at high temperature does not require long-range diffusion of TM ions and avoids problems such as uneven cation distribution and lattice defects. This simple and quick synthesis process is suitable for large-scale promotion and application.
Among them, hydroxide precipitation, especially Mn(OH)2, is easily oxidized in an alkaline environment and an inert protective gas is required to isolate oxygen. However, compared to other carbonates and oxalates, hydroxides have higher reactivity and promote a better crystal structure. During the precipitation process, the nucleation and growth of crystals should be controlled by adjusting the conditions, such as pH and chelating agent. The morphology of the primary and secondary particles formed by aggregation is controlled by adjusting the temperature and stirring speed[79]. During the preparation of the precursor, the sheet morphology can be controlled by adjusting the ammonia concentration, thereby synthesizing particles with specific {010} crystal planes dominantly exposed to promote the rapid diffusion of Li+. Furthermore, primary particles with a radially ordered arrangement can be generated in the final secondary spherical particles by optimizing the ammonia concentration and stirring speed during co-precipitation, so that the radial primary particles of the material undergo a coordinated volume change during circulation to reduce stress, strain and the formation of microcracks. It is also possible to prepare precursor materials with a concentration gradient (or core-shell structure)[114,115] by adding pre-designed metal salt solutions of different concentrations in batches [Figure 11A], thereby preparing gradient materials with special element distributions and better performance. However, it is necessary to pay attention to the the phenomenon that the gradient distribution would be weakened in the high-temperature calcination process.
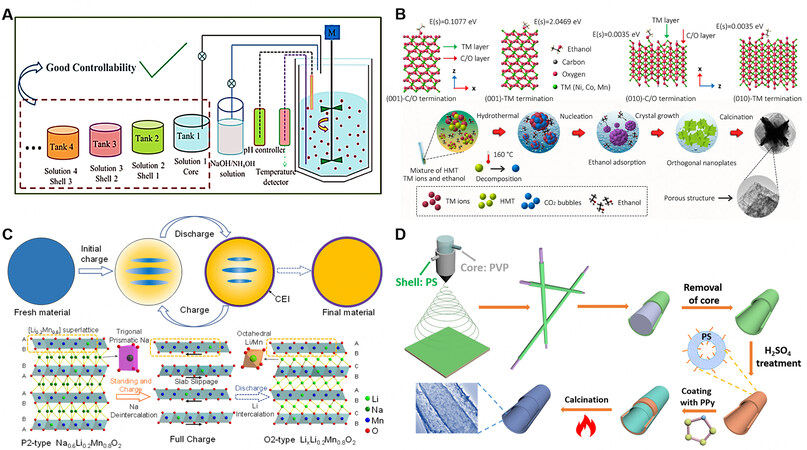
Figure 11. Schematic diagram of (A) materials preparation with gradient distribution of elements by step-by-step co-precipitation method[114]. (B) Controlling nucleation growth to prepare special exposed crystal faces and morphologies by solvothermal method[123]. (C) One-step preparation of O2-type Mn-based cathode material by electrochemical ion exchange[127]. (D) Synthesis process of one-dimensional core-shell structure by electrospinning method[139].
The calcination temperature and time have an important influence on the crystallinity and morphology of a material. The higher the calcination temperature and the longer the hold time, the larger the particle size of the primary particles[116-118], and even the transformation from polycrystalline secondary particles to large single crystal particles occurs, with the structure with a special morphology being more susceptible to damage during the calcination process. It may also cause excessive oxidation of TMs and a loss of Li/O, leading to increased cation mixing and phase transformation[119]. In contrast, a lower calcination temperature and time also reduce the crystallinity of the material. Considering the special morphology and high crystallinity, Chen et al.[18] heated the precursor with the special morphology at a low temperature in advance to pre-oxidize the TM, thereby reducing the reaction temperature and time, while maintaining the crystallinity of the material during the calcination process. The pre-oxidation process can also remove part of CO2 or H2O from the precursor in advance to form a particle morphology with a multilayer hollow core-shell structure. The rate of heating and cooling also affects the morphology and crystal structure of the material. A lower heating rate is more conducive to the maintenance of the particle morphology and a faster cooling method causes more stacking faults, cation disorder and other lattice defects[38].
Solvothermal method
The solvothermal method refers to the rearrangement/recrystallization of ions in the liquid phase at a certain temperature and pressure, which can realize some reactions that are difficult to complete in solid or normal liquid phase reactions. Some new materials with special structures or morphologies can be prepared[120-122]. Specifically, the corresponding molar ratio of metal salt and lithium salt is dissolved in a single or mixed solvent[120] (e.g., deionized water, alcohol, dimethyl formamide and so on) with a certain additive, such as acetic acid[75] and hexamethylenetetramine (HMT)[76]. Subsequently, the prepared solution is transferred to a stainless-steel reactor. The precursor can be collected after reacting at a certain temperature and then calcined at a certain calcination condition to obtain the final material. Among them, the Li source can also be introduced after the solvothermal reaction. The morphology of the materials prepared by the solvothermal method is complex and diverse, including spherical secondary particles, single crystal nanosheets, microrods and 3D layered morphology, formed by orthogonal stacking of nanosheets. Microwave assistance can accelerate the reaction rate in the solvothermal process and increase the pores between the spherical secondary particles for promoting the migration of Li+[77]. The microwave heating method can also be applied to the high-temperature calcination process, which greatly shortens the reaction time.
During the nucleation process, some additives or solvent molecules have the lowest adsorption energy on the {010} crystal plane and tend to be adsorbed on the surface of the crystal plane[123], which results in slower crystal growth in the direction of the crystal plane and promotes the continuous accumulation and growth of atoms along the c-axis direction [Figure 11B]. This promotes the final particle to exhibit more exposed {010} crystal planes. Liu et al.[76] synthesized a series of precursors with a controllable morphology by a solvothermal method, where HMT acts as the precipitant and a mixture of dimethyl formamide and deionized water acts as the solvent. The prepared 3D cube-maze-like Li1.2Mn0.54Co0.13Ni0.13O2 material exhibited excellent Li+ diffusion ability and cycle stability by virtue of its unique 3D structure.
Ion-exchange method
The ion-exchange method means that after the material is placed in the designed solution, a certain ion in the material is spontaneously exchanged with the corresponding ion in the solution and the basic framework of the material is maintained to achieve the preparation of new materials. Among them, the number of ions in the solution must exceed the number of ions in the material to be replaced several times to be completely exchanged. At present, the Li/Na ion exchange reaction is usually used to prepare LIB cathode materials, which can obtain unique structural characteristics based on the precursor with a layered structure[124]. However, the large difference between the Li+ and Na+ radii causes drastic changes in the interlayer spacing and structure of the material, which may be accompanied by partial lattice distortion and particle breakage[125].
The conspicuous O2-type LRMO materials are prepared by molten salt ion exchange based on the precursor of P2-type Na-ion battery cathode materials[126]. Specifically, the P2-type precursor is mixed with an excessive amount of composite lithium salt (LiNO3:LiCl = 88:12 w/w) with a lower eutectic point and then heated to melt the composite lithium salt and cause the Li/Na ion exchange to occur. After a period of the reaction, it is cooled to room temperature, washed clean to remove impurities and the O2-type material is finally obtained. This kind of O2-type structure cannot be obtained with the traditional high-temperature calcination process and can only be obtained using the characteristics of the P2-type material, where the oxygen array slips easily at high potential to generate the O2-type structure.
Furthermore, we have developed an electrochemical ion exchange method[127] based on the working principle of alkaline-ion batteries [Figure 11C]. By assembling the precursor into a hetero-type battery, the ions in the electrolyte and bulk material can be exchanged through the electrochemical driving force by continuous charge and discharge. This method generally needs to circulate the material a certain number of times to achieve a relatively complete exchange. A relatively complete ion exchange will be achieved after certain electrochemical cycles by this method, which is simple to realize one-stop ion exchange and enhanced performance. Ion exchange can also occur when the precursor material is immersed in an organic or inorganic solution[128,129] containing a relatively high concentration and excess of Li+ at room temperature.
Other preparation methods
The sol-gel method[130-132] refers to the stoichiometric ratio of metal salts dissolved in water to prepare a solution, with a certain proportion of complexing agents (e.g., glucose, citric acid and so on) added and heating and stirring to evaporate the solvent. During this process, the metal salt is gradually hydrolyzed or complexed to form a sol and finally dehydrated to form a gel. The gel is dried and calcined to obtain a metal oxide precursor. It is then calcined at a high temperature to obtain the final cathode material after the precursor is uniformly ground. This method can realize the mixing of target ions at the ion/nanoscale, lower the subsequent calcination temperature and is beneficial to realizing the elemental distribution of the cathode material with high uniformity.
The electrospinning method is used to jet-spin the target metal salt polymer solution in a strong electric field to produce a polymer filament precursor with a nanometer diameter and then perform high-temperature sintering to prepare the cathode with one-dimensional nanowires[133-137]. Other petal-shaped nanosheets and nano-block particles can also be prepared by adjusting the sintering temperature and time[138]. In addition, using different preparation processes, such as changing the nozzle structure and controlling the experimental conditions, the precursors with solid, hollow or core-shell structures can be obtained, and finally the preparation of materials with special morphologies or an in-situ coating can be realized [Figure 11D][139].
The spray drying method is used to spray the mixed solution, sol or suspension and other fluid materials that need to be dried under high pressure and dispersed into mist-like droplets. The droplets are sprayed into the drying chamber with fluid hot air, which can instantly remove the moisture and a dry powder particle with uniform mixing of elements will be obtained. The precursor material is then sintered and finally the layered cathode material is prepared. The nanoscale primary particles dispersed in the solution can be spray-dried to produce secondary spherical particles and a more uniform coating can also be achieved by spray drying the coating material[140], such as graphene, and the electrode material. Herein, the properties of different preparation methods and their advantages and disadvantages are prepared in Table 1.
Summary of the properties of different preparation methods and their advantages and disadvantages
| Cost | Process | Production | Morphology | Performance | |
| Co-precipitation method | Low | Simple | Huge | Spheroidal | General |
| Solvothermal method | High | Complex | low | Multiple | Good |
| Ion-exchange method | High | Simple | Huge | Irregular | Good |
| Sol-gel method | Low | Complex | low | Irregular | Good |
| Electrospinning method | High | Complex | low | Fibrous | Good |
| Spray drying method | Low | Simple | Huge | Spheroidal | General |
Modification technology
Despite the in-depth research into LRMOs, their drawbacks still need to be resolved to meet commercial applications. The irreversible O2 release generated near the surface layer after the initial activation of the material poses a severe challenge to the stability and safety of the electrode interface. The destruction of the LiMn6 superlattice and the evolution of the local structure caused by the migration of bulk TMs and oxygen vacancies exert a profound impact on the structure and electrochemical performance. For this reason, a large number of targeted modification measures have been taken for LRMOs to solve the above-mentioned problems.
Regulating activation
When an LRMO is charged to ~4.5 V for the first time, the charge compensation of oxygen occurs, accompanied by the in-plane/out-of-plane migration of TMs and the reorganization of the energy band structure. The irreversible structural changes that partly occur during this process to the subsequent electrochemical behavior and structural evolution are affected profoundly. Watanabe et al.[141] and
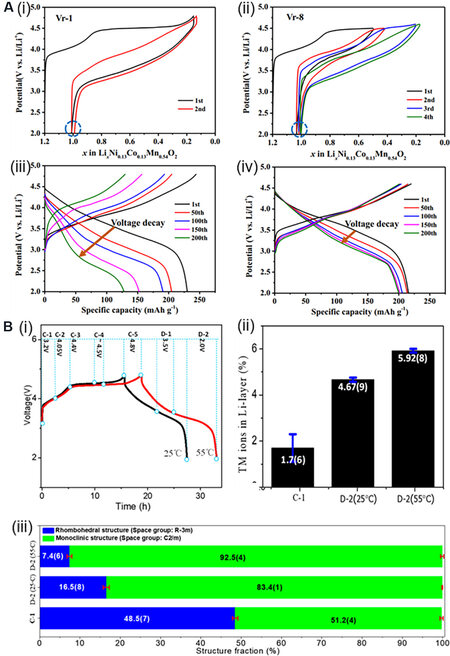
Figure 12. (A) Charge-discharge curves and electrochemical performance of LRMO materials under different voltage systems. (i) Initial two cycles of charge-discharge curves under the condition of Vr-1 (tested between 2.0 and 4.8 V). (ii) Initial four cycles of charge-discharge curves under the condition of Vr-8 (pre-cycled at 2.0 to 4.5 V for two cycles and tested between 2.0 and 4.6 V). (iii and iv) Cycle performance of materials under Vr-1 and Vr-8, respectively[110]. (B) Initial charge-discharge curves (i), the evolution of TM occupancy in Li layer (ii) and two-phase content by Rietveld refinement (iii) for Li1.2Mn0.567Ni0.167Co0.078O2 at 25 and 55 °C[93]. LRMO: Lithium-rich manganese-based layered oxide.
The range of the voltage test is also related to the evolution of electrochemical performance and structure[144]. TMs migrate from the TM layer to the vacancy in the Li layer at a high charging voltage
Kaewmala et al.[149] studied the influence of current density on the structural changes and cycle stability of an LRMO and found that the activation of the Li2MnO3 components was affected by the current density. High-current cycling can effectively reduce the activation of Li2MnO3 and the formation of the spinel phase to obtain better cycling performance and faster Li+ diffusion. Yu et al.[93] studied the influence of activation temperature on the phase structural transformation [Figure 12B] and found that the remaining proportion of the Li2MnO3 component after activation at 25 °C is 17%, while the remaining Li2MnO3 component after activation at 55 °C only remains 7%. This shows that the activation process of LRMOs is very sensitive to temperature and will be aggravated with increasing temperature. Therefore, the activation process, voltage range, current density and other factors are very important for LRMOs. Appropriate working conditions need to be designed to make the electrochemical performance of the material reasonable.
Surface modification
In the process of electrochemical charge-discharge, there are serious problems, such as oxygen release, holes, microcracks, particle powdering, TM dissolution and irreversible phase change of the surface structure, which are the main reasons for the decay of electrochemical properties, so it is necessary to focus on improving the stability of the surface of the material.
Coating is a common means to modify the surface of a material. Coating materials are generally divided into oxides[150-152], phosphates[153], fluorides[154,155] and conductive polymers[156], and the surface treatment object can be an electrode material or precursor. Inert oxides, such as Al2O3[157] and SnO2[158], can only build an inactive isolation layer on the surface of the materials to avoid the side reactions caused by direct contact with the electrolyte, but they may reduce the electron/ion conductivity and affect the rate performance. Other functional oxides enriched with oxygen vacancies, such as Ce0.8Sn0.2O2-σ[159], MoOx[160] and so on, not only avoid direct contact between the host material and the electrolyte, but also provide oxygen vacancies to capture oxygen radicals, a fast ionic conductor reacting with surface Li to improve rate performance[161,162], additional charge compensation to improve the capacity and other functions[163] [Figure 13A]. Fluoride coatings provide F-O bonds on the surface, thus reducing oxygen loss and the decomposition of the electrolyte. Phosphates can also stabilize the surface lattice oxygen to reduce oxygen loss and PO43- can form covalent bonds with metal ions to inhibit TM migration. Conductive polymer coating layers can significantly improve the electrochemical performance due to its high electronic conductivity and good coating uniformity. In addition, the binding force between the surface coating layer and the host material cannot be ignored. It is a good choice to choose a coating that is structurally compatible with a bonding effect[164].
Figure 13. (A) Schematic illustration of oxygen vacancies in Ce0.2Sn0.8O2-σ and its protective effect on an LRMO material[159]. (B) HAADF image near the surface (i) and the EDS mapping of Mn and Nb (ii) of the LRMO particle by Nb surface doping[170]. (C) Schematic of gas-solid interface reaction between Li-rich layered oxides and carbon dioxide[173]. (D) Optimized structure of BO45--doped LRMO (left), in which B atoms occupy the tetrahedral interstitial sites. The simulated local structure (right) around the boracic polyanion, showing that the M-O bonds are lengthened due to the strong B-O bond[182]. (E) Schematic illustration of Li-rich layered material with composited LiMn6 and SbNi6 superstructures[186]. (F) (i) Length of TM-O (Cl) bonds of Li1.2Mn0.53Ni0.27O2 (LMNO) and Li1.2Mn0.53Ni0.27O1.976Cl0.024 (LMNOC), (ii) energy barriers of TM migrating from the TM layer to the Li layer and (iii) operando differential electrochemical mass spectrometry curves of O2 for LMNO and LMNOC[187]. LRMO: Lithium-rich manganese-based layered oxide; HAADF: high-angle annular dark-field.
Surface doping is based on surface coating. By increasing the calcination temperature, the coated elements spontaneously diffuse into the surface layer lattice of the material to form localized interface doping[165-167]. Surface doping can enhance the interaction between the doped elements and the host material and stabilize the surface interface without affecting the bulk structure[168,169]. Liu et al.[170] modified the surface of
In addition, other surface treatments, such as mild acid treatments[172], can achieve H+/Li+ exchange and remove Li+ and O2- in the Li2MnO3 component in advance to reduce oxygen release and irreversible capacity loss during the initial cycle. Simultaneously, the residual Li2CO3, LiOH and other impurity components on the surface of the host material were removed, which improves the reaction activity of the cathode interface. Gas phase[73,173,174] (NH3, CO2, CO and so on) treatment can produce certain element doping or oxygen vacancy generation on the surface of the material, inhibit the oxygen release and promote the Li+ diffusion [Figure 13C]. After molten salts or other highly reactive solutions are used to act on LRMOs[175-177], epitaxial heterostructures[150] (spinel and cation disordered rock-salt phases) are inclined to form on the surface. These structures formed by the phase transformation of layered materials exhibit higher thermodynamic stability, can inhibit the activity of surface oxygen and stabilize the interface to avoid the migration of structural defects from the surface to the bulk.
Bulk doping
Compared with the surface layer, the structural stability and electrochemical reversibility of the bulk phase of the material are relatively good. However, due to its intrinsic anion redox reaction, there are still problems such as TM migration, oxygen vacancy transfer and phase structural transformation. Therefore, bulk doping of the material is expected to improve the electrochemical performance and structural stability of the material radically. Generally, bulk doping can be carried out at three positions: Li sites[178]; TM sites[107]; oxygen active sites[96,179,180], which can produce different effects and results.
Liu et al.[181] prepared Li1.167-xKxMn0.583Ni0.25O2 by ball milling and solid-phase sintering, using K+ to partially replace Li+ in the material. The rate performance and cycle performance of the pristine material are significantly improved. Studies have found that the doped K+ is pinned at the Li site, which hinders the migration path of part of the Mn and inhibits the formation of the spinel phase. Zhang et al.[178] successfully incorporated Na+ into the Li1.23[Ni0.2464Mn0.462Co0.0616]O2 crystal through a solid-phase reaction to partially replace Li+ in bulk. Tests showed that Na+ doping significantly improved the electrochemical performance of the material. The main reason is that Na+ doping into the layered structure expanded the layer spacing, which promoted the rapid diffusion of Li+ and reduced the release of oxygen in the crystal lattice. Meanwhile, Mn4+ in Li2MnO3 was further activated after Na+ doping and provided reversible capacity in subsequent electrochemical cycles.
Li et al.[182] successfully doped boron polyanions into a cathode material to synthesize
Jiang et al.[185] used F- to replace O2- in Li1.2Ni0.2Mn0.6O2, which changed the local electronic environment of Mn, reduced the release of oxygen and caused a change in the surface structure. The symmetry of the MnO6 octahedron is destroyed because of the shorter Mn-F bond than the Mn-O bond, thereby alleviating the Jahn-Teller effect. The expansion of the interlayer spacing after doping caused Li+ to be inserted/extracted faster, thereby suppressing the increase in charge transfer resistance during cycling. In addition, the conversion of the layered structure to the spinel phase during cycling was inhibited, which finally improved the electrochemical performance. Li et al.[186] successfully prepared Li(Li1/6Mn1/3Ni1/3Sb1/6)O2 materials with mixed Li@Mn6 and Sb@Ni6 superstructure elements [Figure 13E]. The study found that the coordination environment of local oxygen changes, which reduces the energy barrier for Li+ diffusion and provides additional capacity. Wang et al.[187] partially replaced oxygen with anion Cl to change the coordination environment of the TMs, which stabilized the TM-O bond and effectively inhibited the irreversible O2 release and TM migration [Figure 13F], thereby enabling the material to exhibit excellent capacity and voltage retention.
Other electrode/electrolyte scope
In order to ensure that an electrode exhibits good electrochemical performance, a certain amount of conductive agent is usually added during the production of the electrode slice to reduce the contact resistance and improve the electronic conductivity. The conductive agent should possess good dispersibility and a high specific surface area and electrolyte adsorption capacity to further improve the ionic conductivity of the electrode. The main conductive additive in traditional cathodes is acetylene black; however, acetylene black is difficult to disperse and exhibits strong oil absorption, resulting in an uneven local composition of the slurry. Yu et al.[188] used carbon nanotubes (CNTs) to prepare composite LRMO cathode electrodes, which displayed the characteristics of a binder, lightweight and self-standing [Figure 14A]. Based on long-period electrochemical tests, the self-supporting structure can reduce polarization, stabilize the electrode to inhibit structural deformation and simultaneously promote the rapid transmission of electrons/ions.
Figure 14. (A) Schematic synthesis process of self-standing CNT-LRMO (i) with photographic (ii) and SEM images (iii)[188]. (B) Chemical structure of GG and PVDF (i), photographs of electrolyte (ii) and GG (iii) and the mixture of GG and electrolyte after 0 h (iv) and 24 h (v)[190]. (C) Schematic of the effect of PAA binder for LRMO electrode[191]. (D) SEM micrograph of PVDF-HFP separator (i-iii) and modified PVDF-HFP/GO nanosheet (iv-vi) in its original state and stored at 150/170 °C for 1 h[192]. CNT-LRMO: Carbon nanotube-lithium-rich manganese-based layered oxide; GG: guar gum; PAA: polyacrylic acid; HFP: hexachloropropylene; GO: graphene oxide.
As an inactive ingredient, the binder cannot provide capacity. Currently, the matching binder for LRMOs is mainly based on a fluorocarbon (PVDF). PVDF is sensitive to the water content during the electrode preparation process and will swell in the electrolyte. Furthermore, its exothermic reaction with metal lithium and LixC6 at higher temperatures is unfavorable from a safety perspective. Therefore, designing and synthesizing a binder with excellent conductivity and viscosity is very important for improving the energy density, rate and cycle performance. Zhang et al.[190] used guar gum (GG) as the binder of a LRMO, which exhibited low solubility and swelling ability in the electrolyte [Figure 14B] and effectively reduced the volume expansion of the electrode during cycling. Polyacrylic acid (PAA) was also used by Yang et al.[191] as a binder for LRMO electrodes [Figure 14C]. The H+/Li+ exchange between the LRMO and PAA generates protons on the surface layer of the cathode, which effectively suppress the migration of TMs, thereby forming a stable structure. This surface modification represents a new method for stabilizing LRMOs.
Currently, microporous polyolefin membranes, such as polyethylene and polypropylene, are the most commonly used separators for lithium-rich cathode materials, because of their excellent mechanical strength and chemical stability. However, the low melting point of polyolefin membranes results in high thermal shrinkage at high temperatures, causing an internal short circuit and even explosion. Simultaneously, the non-polar polyolefin separator exhibits poor wettability, which has a negative impact on the rate and cycle performance of the battery. Choi et al.[192] incorporated graphene oxide (GO) nanosheets into a PVDF-hexachloropropylene (HFP) framework through phase inversion during the pore formation process [Figure 14D]. The addition of GO greatly improves the thermal stability of the composite separator. Shin et al.[193] reported a new concept of coating nitrogen and sulfur co-doped graphene (NSG) nanosheets on a polyethylene separator. The thin NSG layer acts as a physical barrier to inhibit Li dendrite growth due to its excellent mechanical properties. Due to the local strain caused in the carbon skeleton, the incorporation of N and S can cause structural deformation, thereby improving ion migration while maintaining a uniform ion flux on the surface of the Li metal.
The high working voltage of an LRMO exceeding 4.5 V causes the traditional carbonate-based electrolyte[194,195] to decompose severely on the surface of the cathode materials[196], forming an uneven, unstable and unprotected CEI, which hinders the diffusion of Li+ and reduces the electrochemical efficiency. Liu et al.[21] studied the effect of concentrated electrolyte (with a solute concentration of 3 mol L-1) on the surface structure of a cobalt-free LRMO and found that high-concentration electrolytes can form a CEI rich in LiF and other inorganic species on the surface of the electrode, which significantly inhibited the dissolution of TM and effectively protected the cathode material from corrosion. Cui et al.[197] added lithium difluoro(oxalato)borate to a perfluorinated electrolyte and the strong fluorinated CEI formed in situ effectively reduced the side reaction between the O2-type LRMO electrode and electrolyte, and further suppressed O2 structural transformation and oxygen release. Lou et al.[198] first studied methyl diphenyl phosphonate as an electrolyte additive to form a strong artificial mesophase layer in situ on the surface of the cathode electrode. The intermediate phase can effectively inhibit the decomposition of the electrolyte and greatly improve the stability of the interface between Li1.16Ni0.2Co0.1Mn0.54O2 and the electrolyte under high voltage and temperature.
TESTING AND CHARACTERIZATION METHODS
The unique anion charge compensation mechanism of LRMO cathode materials causes a series of complex scientific problems, such as TM migration, oxygen release, surface densification, lattice distortion and energy band reconstruction. It is therefore necessary to employ multiscale and omni-directional characterization methods for in-depth research and effectively carry out materials design to develop new high specific energy cathode materials based on anionic redox reactions.
Electrochemical methods
Galvanostatic charge-discharge tests can act on the electrode material in a certain voltage range and obtain electrochemical properties, such as the charge and discharge curve, capacity, cycle and rate performance, of the material. The charge and discharge curve of the material can be differentiated to obtain dQ/dV, which can identify the redox reaction occurring at different potentials. The electrochemical behavior and evolution of the material can be obtained by analyzing the position and migration of the redox peaks. Yue et al.[199] conducted galvanostatic charge-discharge tests under different voltage ranges. By analyzing and comparing different dQ/dV curves [Figure 15A], the electrode reaction potentials and reaction ranges of different redox couples were easily and effectively obtained.
Figure 15. (A) Different electrochemical redox couples judged by regulating the voltage range. Charge-discharge curves (i) and corresponding dQ/dV (ii) by fixing the lower cut-off voltage and gradually increasing the upper cut-off voltage. Charge-discharge curves (iii) and corresponding dQ/dV (iv) by fixing the upper cut-off voltage and gradually decreasing the lower cut-off voltage[199]. (B) CV curves of different LRMO materials at different scanning rates and the linear fit of the ip and v1/2 in the illustration[201]. (i)
The principle of cyclic voltammetry (CV) is to apply a linearly varying cyclic voltage on a working electrode to record the change in potential. According to the peak current and peak potential in the cycle diagram, the electrochemical reaction of the electrode within this potential range can be analyzed. It can read the reaction type, steps and mechanism, judge the reversibility of the reaction and even study the chemical reaction that occurs on the electrode surface. Yu et al.[200] prepared Ga-doped Li[Li0.2Mn0.54Co0.13Ni0.13-xGax]O2 and used CV to test the material at a scan rate of 0.2 mV s-1 at 2.0-4.8 V to evaluate its electrochemical stability. By comparing the peak position, peak and potential in the initial two circles of the CV curve with the original electrode, the Ga-doped cathode material exhibited a larger reduction peak current, indicating that Ga doping can help improve the Mn4+/Mn3+ reactive activation. The apparent DLi+ of LRMO materials can also be calculated through CV tests with different scanning rates:
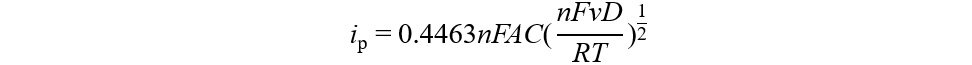
where ip represents the current maximum in amps, n is the number of electrons transferred in the redox process, A is the electrode area, C is the ion concentration, F is the Faraday constant, R is the gas constant, T is the test temperature, v refers to the scan rate of CV and D is the diffusion coefficient [Figure 15B].
EIS is based on the application of a small amplitude sine wave potential (or current) disturbance signal to the electrode, so that the electrode system produces an approximately linear response and information, such as impedance, of the electrode can be obtained through these signals. In a LIB system with a LRMO cathode material, the side reactions of the interface cause the formation and accumulation of electrochemical inert materials on the electrode surface and the structural phase transition also causes changes in the internal resistance of the battery. Therefore, the use of EIS to characterize the diffusion capacity of Li+ inside the material and the surface charge transfer resistance of the material is of great significance. EIS was adopted by Xue et al.[156] to further analyze the influence of polyaniline (PANI)-coated LRMO cathode materials on Li+ diffusion and electron migration. The equivalent circuit model calculation based on the EIS spectrum showed that the surface film resistance of the cathode material coated with PANI decreased and the growth of the charge transfer resistance slowed down, indicating that the electrochemical activity of the interface was significantly improved after coating. The AC impedance method can also use the following formula to calculate the ion diffusion coefficient:
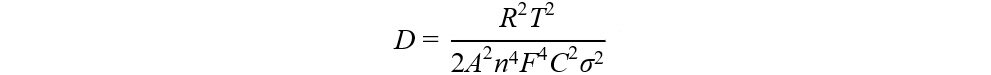
where σ is the Warburg coefficient and the other coefficients are consistent with those in the above CV method [Figure 15C]. Redel et al.[201] systematically studied the electrode reaction kinetics and calculated the DLi+ of different molar amounts of LRMO materials through CV and EIS. They found that
The galvanostatic intermittent titration technique (GITT) is a common method for measuring DLi+ in LIB electrode materials. The principle is to apply a constant current to the electrode for a certain period of time and record and analyze the potential corresponding curve after the current pulse. This method eliminates the ohmic potential drop that is difficult to avoid in methods, such as constant potential intermittent titration techniques, and can quickly calculate the DLi+ of the electrode material at any potential. The formula for calculating the diffusion coefficient is:
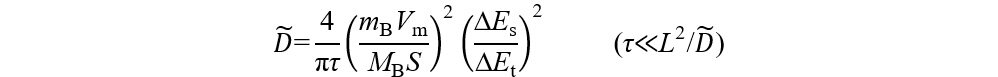
where τ is the time of the current pulse, mB, MB, Vm and S represents the mass, molecular weight, partial molar volume and the surface area of the electrode materials, respectively, and ΔEτ and ΔEs are the voltage changes during the current pulse and standing process, respectively [Figure 15D]. Yang et al.[110] conducted a GITT test on the initial charge and discharge process of the cathode and found that the value dropped sharply at ~4.5 V, thereby confirming the slow kinetics of anion charge transfer in LRMO materials.
Physical characterization
The anion redox mechanism is unique to LRMOs among layered cathode materials. The main test methods introduced in this section are used to characterize the complex charge compensation behavior of oxygen.
Shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS) can obtain information on molecular vibration and rotation and apply it to molecular structure research by analyzing scattering spectra with different frequencies from the incident light. Li et al.[87] successfully detected the peroxygen signal of Li0.66[Li0.12Ni0.15Mn0.73]O2 through the in-situ SHINERS, which confirmed the formation of oxidized lattice oxygen species during the initial two charge-discharge processes [Figure 16A]. In addition, the shift of the related peaks of the oxidized lattice oxygen indicates the contraction of the O-O bond after Li+ deintercalation. When discharged to 2.0 V, the disappeared signal shows that the oxidized oxygen species can be reversibly reduced back to the initial state, which further proves the reversibility of the lattice oxygen redox. Hy et al.[202] also used SHINERS to study the evolution of Li2O in a LRMO and clarified the reaction mechanism of oxygen activation.
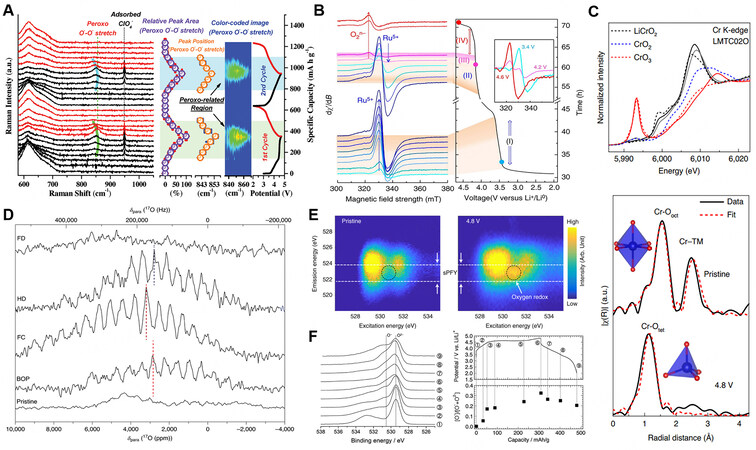
Figure 16. (A) In-situ SHINERS spectra recorded during initial two cycles with reversible generation and migration of peroxo O--O- at high voltage[87]. (B) X-band in-situ EPR spectra of Li2Ru0.75Sn0.25O3 during the initial charge between 2.0 and 4.6 V. Ru4+/5+ charge compensation occurs in the low-voltage plateau area and the reductive coupling of Ru and oxygen occurs in the first half of the high-voltage plateau with the oxidation process of pure O2- in the second half[203]. (C) Cr K-edge XAS spectra (top) of Li1.2Mn0.2Ti0.4Cr0.2O2 in pristine (black solid line), 4.8 V-charged (red solid line) and 1.5 V-discharged states (gray solid line). LiCrO2, CrO2 and CrO3 standard spectra are plotted as dashed lines. Fitted Cr K-edge EXAFS spectra (bottom) of Li1.2Mn0.2Ti0.4Cr0.2O2 in pristine and 4.8 V-charged states. Peaks at ~1.0, ~1.5 and ~2.5 Å correspond to the tetrahedral Cr-O, octahedral Cr-O and Cr-TM coordinations, respectively[109]. (D) Solid-state 17O MAS NMR spectra showing the molecular O2 trapped in the bulk of the cathode particles in the state of full charge[41]. (E) Soft X-ray RIXS data proves that oxygen redox occurs when Li1.2Ni0.13Co0.13Mn0.54O2 is charged to 4.8 V[205]. (F) O 1s HAX-PES spectra (left) of Li[Li0.25Ni0.20Mn0.55]O1.93 electrodes for the 1st cycle. The sampling points correspond to the image on the right with the intensity ratio of [O-]/[O- + O2-][207]. SHINERS: Shell-isolated nanoparticle-enhanced Raman spectroscopy; EPR: electron paramagnetic resonance; XAS: X-ray absorption spectroscopy; EXAFS: extended X-ray absorption fine structure; NMR: nuclear magnetic resonance; RIXS: resonant inelastic X-ray scattering; HAX-PES: hard X-ray photoelectron spectroscopy.
Electron paramagnetic resonance (EPR) can be used to qualitatively and quantitatively detect the unpaired electrons contained in the atoms or molecules of substances and to explore the structural characteristics of the surrounding environment. Sathiya et al.[203] reported the results of the EPR operation spectroscopy and in-situ imaging of Li2Ru0.75Sn0.25O3 [Figure 16B]. By operando monitoring of the EPR signal of Ru5+ and paramagnetic oxygen, the formation of reversible O2n- species can be clearly proved. Tang et al.[86] used EPR to in-situ detect the cation-anion redox correlation and found that the coupling between Mn and oxygen produces the necessary synergy to stabilize O2n- species, activates more Mn to participate in the electrochemical reaction and ensures the high specific capacity of the LRMO material.
The structural information in the extended X-ray absorption fine structure (EXAFS) spectrum comes from the interference between the photoelectron wavefunction and nearby atoms. The structural information, such as the coordination structure around different element atoms and the types of coordination atoms, contained in the spectrum can be interpreted using Fourier transform. Huang et al.[109] analyzed the coordination environment of Cr in lithium-rich disordered rock salt phase materials by EXAFS spectroscopy and found that during the charging process, Cr migrates from the initial octahedral position to the tetrahedral coordination environment [Figure 16C]. This non-topological reaction mechanism promotes the transformation of the Li+ migration path from the 1-TM to 0-TM channel and realizes rapid Li+ diffusion. After analyzing the EXAFS spectrum of the TM and oxygen in different charge-discharge stages, Yin et al.[71] revealed the evolution of their coordination structure and studied the effect of different nickel contents on anion redox. They showed that an appropriate increase in Ni content not only changed the redox center of the LRMO cathode but also enhanced the reversibility of Mn migration through the shielding effect.
NMR is one of the most powerful tools to qualitatively analyze the composition and structure of materials through the absorption of radio-frequency radiation by the atomic nucleus and sometimes can also be used for quantitative analysis. In order to independently verify the formation and reduction of molecular O2, House et al.[41] labeled Li1.2Ni0.13Co0.13Mn0.54O2 samples with 17O. The results showed a large shift in the peak position and an 17O paramagnetic shift. In addition, the 17O sideband shift composed of a series of spikes indicates that the molecular O2 is strictly confined in its local environment [Figure 16D], consistent with the clustering of defect vacancies and clearly explaining the formation of molecular O2 and its position in bulk LRMO.
RIXS provides information regarding the intrinsic excitation of materials by detecting the energy and momentum differences of scattered photons. It is mostly used to study the conduction band, valence band and low-energy excited state, as well as the electronic structure of the electronic transition and charge transfer information. Xu et al.[204] used RIXS technology to reveal the attenuation information of soft XAS excitation energy, which provided higher sensitivity and clarity for defining the chemical state of oxygen and explained the charge compensation mechanism of the TMs in two compounds (Li1.2Ni0.2Mn0.6O2 and Li1.2Ni0.2Ru0.6O2). Simultaneously, Zhang et al.[205] applied RIXS to find that O-1 was formed in the subsurface transition layer of the material when charged to a high voltage [Figure 16E]. The irreversible loss of oxygen during the charging process decreases the valence of the TM cations and leave the oxygen vacancies around simultaneously, explaining why the valence state of TMs in the LRMO is different from the traditional NCM material. Dai et al.[206] distinguished the redox process of lattice oxygen and TM through mRIXS, quantified the contribution of lattice oxygen redox reaction to electrochemical capacity and revealed the inner behavior of oxygen redox in LRMOs.
Hard X-ray photoelectron spectroscopy (HAX-PES) uses high-energy hard X-rays as the excitation source (such as Cr Kα, energy 5414.9 eV, or a synchrotron radiation source), which can obtain several times the depth of conventional XPS analysis. The chemical information greatly reduces the interference from the surface contamination layer and can analyze the material interface layer non-destructively. It is a very important characterization method for the surface/interface chemical analysis of solid materials.
SUMMARY AND OUTLOOK
Anion charge compensation contributes to the ultra-high specific capacity and energy density of LRMOs; however, it is accompanied by problems, such as capacity attenuation and voltage decay, derived from complex and rapid phase structural evolution. It is also noteworthy that the intrinsic initial Coulombic efficiency and sluggish electrochemical dynamics cannot be ignored. In the past two decades, researchers have devoted significant efforts to studying the key scientific issues behind these problems, as well as to improving electrochemical performance through various optimization methods, with impressive progress having been made. However, there are still a series of challenges hindering the further development and commercialization of LRMOs:
(1) Current detection methods are still unable to in-situ non-destructively monitor the evolution of materials at the electronic/atomic scale during charge and discharge. It is necessary to develop deeper and more accurate detection methods to obtain clearer structural information and verify the results of theoretical simulations. More importantly, powerful and versatile characterization methods need to be developed.
(2) The modified results of the materials are varied, although the electrochemical performance of some materials has improved significantly, the performance of the contrast samples may be very poor.
(3) Although O2-type lithium-rich materials can significantly inhibit the voltage attenuation caused by TM migration and exhibit ultra-high specific capacity, the preparation method of molten salt ion exchange is cumbersome and costly and is therefore difficult to realize practical applications.
(4) Components, such as electrolytes, binders and conductive agents, compatible with LRMOs still need to improve their high-voltage tolerance and coordination.
For pristine LRMOs, the Li/O and the ratio between TM ions should be adjusted, the advantages and defects of each chemical ratio of the material should be clarified clearly. Through electrochemical regulation of voltage and current, select suitable materials to meet the needs of different working conditions. For modification methods, such as doping and coating, it is necessary to focus on the uniformity and consistency of the modified material and eliminate the interference of human factors and randomness during the characterization process. The regulation of highly active near-surface structure should be emphatically considered. The LiMn6 honeycomb superstructure triggers anion charge compensation and voltage hysteresis, so it is necessary to seek effective element substitution or structural adjustment (such as ribbon-like and mesh-like superstructures)[209,210] to achieve stable superstructure and reversible oxygen redox. For the voltage attenuation caused by the irreversible migration of TM to the Li layer, it is vital to focus on suppressing the migration of TM from the transitional state tetrahedral position to the final state octahedral Li vacancy. The O2 type structure is a good choice, but the preparation method still needs to be improved. Other oxygen stacking design[128,211] (such as T#2/O4/O6 type, stacking fault and so on) are also worthy of discussion. For other components, such as electrolyte and binder, functional design is required to stabilize the interface of lithium-rich materials and suppress battery flatulence and safety hazards caused by oxygen release.
Energy density is an eternal goal and the development of commercial cathode materials has been relatively slow. As the embedded cathode materials for LIBs with the highest energy density at present, LRMOs are the most promising next-generation cathode materials for popularization and application in the near future. Compared with NCM materials, LRMOs exhibit considerable advantages in cost and thermal stability but with similar preparation method and equipment. Practical applications can be attempted under the conditions of high energy density and less cycle number requirements. Of course, safety issues caused by high voltage and oxygen release should be considered. The unique oxygen charge compensation mechanism has opened a new field for the realization of high energy density LIBs. It can also provide a reference for other alkali ion batteries, such as Na+/K+ and even Li-S, Li-O2 and other energy storage systems with anion charge compensation. Promoting continuous development of LIBs and even electrochemical energy devices requires constant efforts and exploration from scientific researchers.
DECLARATIONS
Authors’ contributions
Investigation, writing-original draft, review & editing: Yang Z
Investigation: Zheng C, Wei Z, Liu H, Feng J
Language check: Zhong J
Supervision funding supporting: Li J
Supervision: Kang F
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work is financially supported by the National Natural Science Foundation of China (Grants 51972023).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2022.
REFERENCES
1. Choi C, Ashby DS, Butts DM, et al. Achieving high energy density and high power density with pseudocapacitive materials. Nat Rev Mater 2020;5:5-19.
2. Nitta N, Wu F, Lee JT, Yushin G. Li-ion battery materials: present and future. Mater Today 2015;18:252-64.
4. Liu C, Neale ZG, Cao G. Understanding electrochemical potentials of cathode materials in rechargeable batteries. Mater Today 2016;19:109-23.
5. Assat G, Tarascon J. Fundamental understanding and practical challenges of anionic redox activity in Li-ion batteries. Nat Energy 2018;3:373-86.
6. Moghadam Y, Masoudpanah S, Alamolhoda S, Daneshtalab R. Electrochemical properties of LiMn1.5Ni0.5O4 powders synthesized by solution combustion method: Effect of CTAB as a fuel. Advanced Powder Technology 2020;31:639-44.
7. Moghadam Y, El Kharbachi A, Diemant T, Melinte G, Hu Y, Fichtner M. Toward better stability and reversibility of the Mn4+/Mn2+ double redox activity in disordered rocksalt oxyfluoride cathode materials. Chem Mater 2021;33:8235-47.
8. Hu S, Pillai AS, Liang G, et al. Li-rich layered oxides and their practical challenges: recent progress and perspectives. Electrochem Energ Rev 2019;2:277-311.
9. Thackeray MM, Kang S, Johnson CS, Vaughey JT, Benedek R, Hackney SA. Li2MnO3-stabilized LiMO2 (M = Mn, Ni, Co) electrodes for lithium-ion batteries. J Mater Chem 2007;17:3112.
10. Sathiya M, Rousse G, Ramesha K, et al. Reversible anionic redox chemistry in high-capacity layered-oxide electrodes. Nat Mater 2013;12:827-35.
11. McCalla E, Abakumov AM, Saubanère M, et al. Visualization of O-O peroxo-like dimers in high-capacity layered oxides for Li-ion batteries. Science 2015;350:1516-21.
12. Zhou YN, Ma J, Hu E, et al. Tuning charge-discharge induced unit cell breathing in layer-structured cathode materials for lithium-ion batteries. Nat Commun 2014;5:5381.
13. Li B, Jiang N, Huang W, Yan H, Zuo Y, Xia D. Thermodynamic Activation of charge transfer in anionic redox process for Li-Ion batteries. Adv Funct Mater 2018;28:1704864.
14. House RA, Marie J, Pérez-osorio MA, Rees GJ, Boivin E, Bruce PG. The role of O2 in O-redox cathodes for Li-ion batteries. Nat Energy 2021;6:781-9.
15. Hu E, Yu X, Lin R, et al. Evolution of redox couples in Li- and Mn-rich cathode materials and mitigation of voltage fade by reducing oxygen release. Nat Energy 2018;3:690-8.
16. Lei Y, Ni J, Hu Z, et al. Surface modification of Li-rich Mn-based layered oxide cathodes: challenges, materials, methods, and characterization. Adv Energy Mater 2020;10:2002506.
17. Sun Z, Xu L, Dong C, et al. Enhanced cycling stability of boron-doped lithium-rich layered oxide cathode materials by suppressing transition metal migration. J Mater Chem A 2019;7:3375-83.
18. Chen L, Su Y, Chen S, et al. Hierarchical Li1.2Ni0.2Mn0.6O2 nanoplates with exposed {010} planes as high-performance cathode material for lithium-ion batteries. Adv Mater 2014;26:6756-60.
19. Sun G, Yu F, Que L, et al. Local electronic structure modulation enhances operating voltage in Li-rich cathodes. Nano Energy 2019;66:104102.
20. Hwang J, Myeong S, Lee E, et al. Lattice-oxygen-stabilized Li- and Mn-rich cathodes with sub-micrometer particles by modifying the excess-Li distribution. Adv Mater 2021;33:e2100352.
21. Liu W, Li J, Li W, Xu H, Zhang C, Qiu X. Inhibition of transition metals dissolution in cobalt-free cathode with ultrathin robust interphase in concentrated electrolyte. Nat Commun 2020;11:3629.
22. Ji X, Xia Q, Xu Y, Feng H, Wang P, Tan Q. A review on progress of lithium-rich manganese-based cathodes for lithium ion batteries. J Power Sources 2021;487:229362.
23. Cui S, Gao M, Li G, Gao X. Insights into Li-rich Mn-based cathode materials with high capacity: from dimension to lattice to atom. Adv Energy Mater 2022;12:2003885.
24. He W, Guo W, Wu H, et al. Challenges and recent advances in high capacity Li-rich cathode materials for high energy density lithium-ion batteries. Adv Mater 2021;33:e2005937.
25. Seo DH, Lee J, Urban A, Malik R, Kang S, Ceder G. The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Li-excess cathode materials. Nat Chem 2016;8:692-7.
26. Wang Q, Mariyappan S, Rousse G, et al. Unlocking anionic redox activity in O3-type sodium 3d layered oxides via Li substitution. Nat Mater 2021;20:353-61.
27. Song J, Yoon G, Kim B, et al. Anionic redox activity regulated by transition metal in lithium-rich layered oxides. Adv Energy Mater ; doi: 10.1002/aenm.202001207.
28. Zhuo Z, Dai K, Wu J, et al. Distinct oxygen redox activities in Li2MO3 (M = Mn, Ru, Ir). ACS Energy Lett 2021;6:3417-24.
29. Hong J, Gent WE, Xiao P, et al. Metal-oxygen decoordination stabilizes anion redox in Li-rich oxides. Nat Mater 2019;18:256-65.
30. Zhang K, Jiang Z, Ning F, et al. Metal-ligand π interactions in lithium-rich Li2RhO3 cathode material activate bimodal anionic redox. Adv Energy Mater 2021;11:2100892.
31. Yu Z, Qu X, Dou A, Su M, Liu Y, Wu F. Synthesis and redox mechanism of cation-disordered, rock-salt cathode-material Li-Ni-Ti-Nb-O compounds for a Li-Ion battery. ACS Appl Mater Interfaces 2019;11:35777-87.
32. Xie Y, Saubanère M, Doublet M. Requirements for reversible extra-capacity in Li-rich layered oxides for Li-ion batteries. Energy Environ Sci 2017;10:266-74.
33. Ouyang C, Shi S, Lei M. Jahn-Teller distortion and electronic structure of LiMn2O4. J Alloys Compd 2009;474:370-4.
34. Liu S, Wang B, Zhang X, Zhao S, Zhang Z, Yu H. Reviving the lithium-manganese-based layered oxide cathodes for lithium-ion batteries. Matter 2021;4:1511-27.
35. Jarvis KA, Deng Z, Allard LF, Manthiram A, Ferreira PJ. Atomic structure of a lithium-rich layered oxide material for lithium-ion batteries: evidence of a solid solution. Chem Mater 2011;23:3614-21.
36. Yu X, Lyu Y, Gu L, et al. Understanding the rate capability of high-energy-density Li-rich layered Li1.2Ni0.15Co0.1Mn0.55O2 cathode materials. Adv Energy Mater 2014;4:1300950.
37. Yu H, Ishikawa R, So YG, et al. Direct atomic-resolution observation of two phases in the Li(1.2)Mn(0.567)Ni(0.166)Co(0.067)O2 cathode material for lithium-ion batteries. Angew Chem Int Ed Engl 2013;52:5969-73.
38. Xiao L, Xiao J, Yu X, et al. Effects of structural defects on the electrochemical activation of Li2MnO3. Nano Energy 2015;16:143-51.
39. Sun Y, Cong H, Zan L, Zhang Y. Oxygen vacancies and stacking faults introduced by low-temperature reduction improve the electrochemical properties of Li2MnO3 nanobelts as lithium-ion battery cathodes. ACS Appl Mater Interfaces 2017;9:38545-55.
40. Shunmugasundaram R, Arumugam RS, Dahn JR. A study of stacking faults and superlattice ordering in some Li-rich layered transition metal oxide positive electrode materials. J Electrochem Soc 2016;163:A1394-400.
41. House RA, Rees GJ, Pérez-osorio MA, et al. First-cycle voltage hysteresis in Li-rich 3d cathodes associated with molecular O2 trapped in the bulk. Nat Energy 2020;5:777-85.
42. Zhuo H, Liu Y, Wang Z, et al. Insight of reaction mechanism and anionic redox behavior for Li-rich and Mn-based oxide materials from local structure. Nano Energy 2021;83:105812.
43. Zhang J, Cheng F, Chou S, et al. Tuning oxygen redox chemistry in Li-rich Mn-based layered oxide cathodes by modulating cation arrangement. Adv Mater 2019;31:e1901808.
44. Song J, Li B, Chen Y, et al. A high-performance Li-Mn-O Li-rich cathode material with rhombohedral symmetry via intralayer Li/Mn disordering. Adv Mater 2020;32:e2000190.
45. Ning F, Li B, Song J, et al. Inhibition of oxygen dimerization by local symmetry tuning in Li-rich layered oxides for improved stability. Nat Commun 2020;11:4973.
46. Li X, Qiao Y, Guo S, Jiang K, Ishida M, Zhou H. A new type of Li-rich rock-salt oxide Li2Ni1/3Ru2/3O3 with reversible anionic redox chemistry. Adv Mater 2019;31:e1807825.
47. Liu S, Liu Z, Shen X, et al. Li-Ti cation mixing enhanced structural and performance stability of Li-rich layered oxide. Adv Energy Mater 2019;9:1901530.
48. Li N, Sun M, Kan WH, et al. Layered-rocksalt intergrown cathode for high-capacity zero-strain battery operation. Nat Commun 2021;12:2348.
49. Zhao E, Li Q, Meng F, et al. Stabilizing the oxygen lattice and reversible oxygen redox chemistry through structural dimensionality in lithium-rich cathode oxides. Angew Chem Int Ed Engl 2019;58:4323-7.
50. Lee J, Yang Y, Jeong M, et al. Superior rate capability and cycling stability in partially cation-disordered co-free Li-rich layered materials enabled by an initial activation process. Chem Mater 2021;33:5115-26.
51. Liang C, Kong F, Longo RC, et al. Unraveling the origin of instability in Ni-rich LiNi1-2xCoxMnxO2 (NCM) cathode materials. J Phys Chem C 2016;120:6383-93.
52. Zheng J, Gu M, Genc A, et al. Mitigating voltage fade in cathode materials by improving the atomic level uniformity of elemental distribution. Nano Lett 2014;14:2628-35.
53. Liu Q, Zhu H, Liu J, et al. High-performance lithium-rich layered oxide material: effects of preparation methods on microstructure and electrochemical properties. Materials (Basel) 2020;13:334.
54. Wu T, Liu X, Zhang X, et al. Full concentration gradient-tailored Li-rich layered oxides for high-energy lithium-ion batteries. Adv Mater 2021;33:e2001358.
56. Xu CL, Xiang W, Wu ZG, et al. Constructing a protective pillaring layer by incorporating gradient Mn4+ to stabilize the surface/interfacial structure of LiNi0.815Co0.15Al0.035O2 cathode. ACS Appl Mater Interfaces 2018;10:27821-30.
57. Zhu Z, Yu D, Yang Y, et al. Gradient Li-rich oxide cathode particles immunized against oxygen release by a molten salt treatment. Nat Energy 2019;4:1049-58.
58. Yang J, Xia Y. Suppressing the phase transition of the layered Ni-rich oxide cathode during high-voltage cycling by introducing low-content Li2MnO3. ACS Appl Mater Interfaces 2016;8:1297-308.
59. Song Y, Zhao X, Wang C, et al. Insight into the atomic structure of Li2MnO3 in Li-rich Mn-based cathode materials and the impact of its atomic arrangement on electrochemical performance. J Mater Chem A 2017;5:11214-23.
60. Liu J, Liu J, Wang R, Xia Y. Degradation and structural evolution of xLi2MnO3·(1-x)LiMn1/3Ni1/3Co1/3O2 during cycling. J Electrochem Soc 2013;161:A160-7.
61. Shi JL, Xiao DD, Ge M, et al. High-capacity cathode material with high voltage for Li-ion batteries. Adv Mater 2018;30:1705575.
62. Shen S, Hong Y, Zhu F, et al. Tuning electrochemical properties of Li-rich layered oxide cathodes by adjusting Co/Ni ratios and mechanism investigation using in situ X-ray diffraction and online continuous flow differential electrochemical mass spectrometry. ACS Appl Mater Interfaces 2018;10:12666-77.
63. Hua W, Wang S, Knapp M, et al. Structural insights into the formation and voltage degradation of lithium- and manganese-rich layered oxides. Nat Commun 2019;10:5365.
64. Sun G, Yu FD, Zhao C, et al. Decoupling the voltage hysteresis of Li-rich cathodes: electrochemical monitoring, modulation anionic redox chemistry and theoretical verifying. Adv Funct Mater 2020;31:2002643.
65. Zhou Y, Cui H, Qiu B, et al. Sufficient oxygen redox activation against voltage decay in Li-rich layered oxide cathode materials. ACS Materials Lett 2021;3:433-41.
66. Wu F, Tian J, Su Y, et al. Effect of Ni(2+) content on lithium/nickel disorder for Ni-rich cathode materials. ACS Appl Mater Interfaces 2015;7:7702-8.
67. Ji H, Wu J, Cai Z, et al. Ultrahigh power and energy density in partially ordered lithium-ion cathode materials. Nat Energy 2020;5:213-21.
68. Boivin E, Guerrini N, House RA, et al. The role of Ni and Co in suppressing O-loss in Li-rich layered cathodes. Adv Funct Mater 2021;31:2003660.
69. Huang W, Lin C, Zhang M, et al. Revealing roles of Co and Ni in Mn-rich layered cathodes. Adv Energy Mater 2021;11:2102646.
70. Xiang X, Knight JC, Li W, Manthiram A. Understanding the effect of Co3+ substitution on the electrochemical properties of lithium-rich layered oxide cathodes for lithium-ion batteries. J Phys Chem C 2014;118:21826-33.
71. Yin C, Wan L, Qiu B, et al. Boosting energy efficiency of Li-rich layered oxide cathodes by tuning oxygen redox kinetics and reversibility. Energy Storage Mater 2021;35:388-99.
72. Okubo M, Mizuno Y, Yamada H, et al. Fast Li-Ion insertion into nanosized LiMn(2)O(4) without domain boundaries. ACS Nano 2010;4:741-52.
73. Peng H, Zhao SX, Huang C, Yu LQ, Fang ZQ, Wei GD. In situ construction of spinel coating on the surface of a lithium-rich manganese-based single crystal for inhibiting voltage fade. ACS Appl Mater Interfaces 2020;12:11579-88.
74. Su Y, Chen G, Chen L, et al. Exposing the {010} planes by oriented self-assembly with nanosheets to improve the electrochemical performances of Ni-rich Li[Ni0.8Co0.1Mn0.1]O2 microspheres. ACS Appl Mater Interfaces 2018;10:6407-14.
75. Wei GZ, Lu X, Ke FS, et al. Crystal habit-tuned nanoplate material of Li[Li1/3-2x/3NixMn2/3-x/3]O2 for high-rate performance lithium-ion batteries. Adv Mater 2010;22:4364-7.
76. Liu Y, Wang J, Wu J, et al. 3D cube-maze-like Li-rich layered cathodes assembled from 2D porous nanosheets for enhanced cycle stability and rate capability of lithium-ion batteries. Adv Energy Mater 2019;10:1903139.
77. Shi S, Wang T, Cao M, Wang J, Zhao M, Yang G. Rapid self-assembly spherical Li1.2Mn0.56Ni0.16Co0.08O2 with improved performances by microwave hydrothermal method as cathode for lithium-ion batteries. ACS Appl Mater Interfaces 2016;8:11476-87.
78. Trevisanello E, Ruess R, Conforto G, Richter FH, Janek J. Polycrystalline and single crystalline NCM cathode materials-quantifying particle cracking, active surface area, and lithium diffusion. Adv Energy Mater 2021;11:2003400.
79. Xu X, Huo H, Jian J, et al. Radially oriented single-crystal primary nanosheets enable ultrahigh rate and cycling properties of LiNi0.8Co0.1Mn0.1O2 cathode material for lithium-ion batteries. Adv Energy Mater 2019;9:1803963.
80. Csernica PM, Kalirai SS, Gent WE, et al. Persistent and partially mobile oxygen vacancies in Li-rich layered oxides. Nat Energy 2021;6:642-52.
81. Koga H, Croguennec L, Ménétrier M, et al. Operando X-ray absorption study of the redox processes involved upon cycling of the Li-rich layered oxide L1.20 Mn0.54Co0.13Ni0.13O2 in Li ion batteries. J Phys Chem C 2014;118:5700-9.
82. Nakayama K, Ishikawa R, Kobayashi S, Shibata N, Ikuhara Y. Dislocation and oxygen-release driven delithiation in Li2MnO3. Nat Commun 2020;11:4452.
83. Luo K, Roberts MR, Hao R, et al. Charge-compensation in 3d-transition-metal-oxide intercalation cathodes through the generation of localized electron holes on oxygen. Nat Chem 2016;8:684-91.
84. Yabuuchi N, Yoshii K, Myung ST, Nakai I, Komaba S. Detailed studies of a high-capacity electrode material for rechargeable batteries, Li2MnO3-LiCo(1/3)Ni(1/3)Mn(1/3)O2. J Am Chem Soc 2011;133:4404-19.
85. Strehle B, Kleiner K, Jung R, et al. The role of oxygen release from Li- and Mn-rich layered oxides during the first cycles investigated by on-line electrochemical mass spectrometry. J Electrochem Soc 2016;164:A400-6.
86. Tang M, Dalzini A, Li X, et al. Operando EPR for Simultaneous monitoring of anionic and cationic redox processes in Li-rich metal oxide cathodes. J Phys Chem Lett 2017;8:4009-16.
87. Li X, Qiao Y, Guo S, et al. Direct visualization of the reversible O2-/O- redox process in Li-rich cathode materials. Adv Mater 2018;30:e1705197.
88. Gent WE, Lim K, Liang Y, et al. Coupling between oxygen redox and cation migration explains unusual electrochemistry in lithium-rich layered oxides. Nat Commun 2017;8:2091.
89. Liu H, Chen Y, Hy S, et al. Operando lithium dynamics in the Li-rich layered oxide cathode material via neutron diffraction. Adv Energy Mater 2016;6:1502143.
90. Mohanty D, Sefat AS, Kalnaus S, et al. Investigating phase transformation in the Li1.2Co0.1Mn0.55Ni0.15O2 lithium-ion battery cathode during high-voltage hold (4.5 V) via magnetic, X-ray diffraction and electron microscopy studies. J Mater Chem A 2013;1:6249.
91. Xu B, Fell CR, Chi M, Meng YS. Identifying surface structural changes in layered Li-excess nickel manganese oxides in high voltage lithium ion batteries: a joint experimental and theoretical study. Energy Environ Sci 2011;4:2223.
92. Gao Y, Ma J, Wang Z, Lu G, Chen L. Vacancy-induced MnO6 distortion and its impacts on structural transition of Li2MnO3. Phys Chem Chem Phys 2017;19:7025-31.
93. Yu H, So YG, Ren Y, et al. Temperature-sensitive structure evolution of lithium-manganese-rich layered oxides for lithium-ion batteries. J Am Chem Soc 2018;140:15279-89.
94. Radin MD, Vinckeviciute J, Seshadri R, Van der Ven A. Manganese oxidation as the origin of the anomalous capacity of Mn-containing Li-excess cathode materials. Nat Energy 2019;4:639-46.
95. Kleiner K, Strehle B, Baker AR, et al. Origin of high capacity and poor cycling stability of Li-rich layered oxides: a long-duration in situ synchrotron powder diffraction study. Chem Mater 2018;30:3656-67.
96. Cao S, Wu C, Xie X, et al. Suppressing the voltage decay based on a distinct stacking sequence of oxygen atoms for Li-rich cathode materials. ACS Appl Mater Interfaces 2021;13:17639-48.
97. Zuo Y, Li B, Jiang N, et al. A high-capacity O2-type Li-rich cathode material with a single-layer Li2MnO3 superstructure. Adv Mater 2018;30:e1707255.
98. Eum D, Kim B, Kim SJ, et al. Voltage decay and redox asymmetry mitigation by reversible cation migration in lithium-rich layered oxide electrodes. Nat Mater 2020;19:419-27.
99. Ma J, Zhou Y, Gao Y, et al. Feasibility of using Li2MoO3 in constructing Li-rich high energy density cathode materials. Chem Mater 2014;26:3256-62.
100. Grenier A, Kamm GE, Li Y, Chung H, Meng YS, Chapman KW. Nanostructure transformation as a signature of oxygen redox in Li-rich 3d and 4d cathodes. J Am Chem Soc 2021;143:5763-70.
101. Yan P, Zheng J, Tang ZK, et al. Injection of oxygen vacancies in the bulk lattice of layered cathodes. Nat Nanotechnol 2019;14:602-8.
102. Lin R, Hu E, Liu M, et al. Anomalous metal segregation in lithium-rich material provides design rules for stable cathode in lithium-ion battery. Nat Commun 2019;10:1650.
103. Lee S, Jin W, Kim SH, et al. Oxygen vacancy diffusion and condensation in lithium-ion battery cathode materials. Angew Chem Int Ed Engl 2019;58:10478-85.
104. Xiao B, Liu H, Chen N, et al. Size-mediated recurring spinel sub-nanodomains in Li- and Mn-rich layered cathode materials. Angew Chem Int Ed Engl 2020;59:14313-20.
105. Zheng J, Xu P, Gu M, et al. Structural and chemical evolution of Li- and Mn-rich layered cathode material. Chem Mater 2015;27:1381-90.
106. Zheng J, Ye Y, Liu T, et al. Ni/Li disordering in layered transition metal oxide: electrochemical impact, origin, and control. Acc Chem Res 2019;52:2201-9.
107. Liu P, Zhang H, He W, et al. Lithium deficiencies engineering in Li-rich layered oxide Li1.098Mn0.533Ni0.113Co0.138O2 for high-stability cathode. J Am Chem Soc 2019;141:10876-82.
108. Yang M, Jin J, Shen Y, Sun S, Zhao X, Shen X. Cation-disordered lithium-excess Li-Fe-Ti oxide cathode materials for enhanced Li-ion storage. ACS Appl Mater Interfaces 2019;11:44144-52.
109. Huang J, Zhong P, Ha Y, et al. Non-topotactic reactions enable high rate capability in Li-rich cathode materials. Nat Energy 2021;6:706-14.
110. Yang Z, Zhong J, Liu Y, Li Z, Li J, Yang K. Unveiling the effect of voltage regulation system on the structure and electrochemical properties of lithium-rich cathode materials. J Electrochem Soc 2019;166:A1481-9.
111. Nayak PK, Grinblat J, Levi M, Aurbach D. Electrochemical and structural characterization of carbon coated Li1.2Mn0.56Ni0.16Co0.08O2 and Li1.2Mn0.6Ni0.2O2 as cathode materials for Li-ion batteries. Electrochimica Acta 2014;137:546-56.
112. Li N, Wu J, Hwang S, et al. Enabling facile anionic kinetics through cationic redox mediator in Li-rich layered cathodes. ACS Energy Lett 2020;5:3535-43.
113. Saha S, Assat G, Sougrati MT, et al. Exploring the bottlenecks of anionic redox in Li-rich layered sulfides. Nat Energy 2019;4:977-87.
114. Hou P, Li F, Sun Y, Li H, Xu X, Zhai T. Multishell precursors facilitated synthesis of concentration-gradient nickel-rich cathodes for long-life and high-rate lithium-ion batteries. ACS Appl Mater Interfaces 2018;10:24508-15.
115. Sun YK, Myung ST, Park BC, Prakash J, Belharouak I, Amine K. High-energy cathode material for long-life and safe lithium batteries. Nat Mater 2009;8:320-4.
116. Koga H, Croguennec L, Ménétrier M, Mannessiez P, Weill F, Delmas C. Different oxygen redox participation for bulk and surface: a possible global explanation for the cycling mechanism of Li1.20Mn0.54Co0.13Ni0.13O2. J Power Sources 2013;236:250-8.
117. Nie L, Liang C, Chen S, et al. Improved electrochemical performance of Li-rich layered oxide cathodes enabled by a two-step heat treatment. ACS Appl Mater Interfaces 2021;13:13281-8.
118. Liu J, Chen H, Xie J, Sun Z, Wu N, Wu B. Electrochemical performance studies of Li-rich cathode materials with different primary particle sizes. J Power Sources 2014;251:208-14.
119. Liu Y, Zhuo H, Yin Y, Lu S, Wang Z, Zhuang W. Remaining Li-content dependent structural evolution during high temperature re-heat treatment of quantitatively delithiated Li-rich cathode materials with surface defect-spinel phase. ACS Appl Mater Interfaces 2020;12:27226-40.
120. Cui SL, Zhang X, Wu XW, et al. Understanding the structure-performance relationship of lithium-rich cathode materials from an oxygen-vacancy perspective. ACS Appl Mater Interfaces 2020;12:47655-66.
121. Li Y, Feng X, Cui S, Shi Q, Mi L, Chen W. From α-NaMnO2 to crystal water containing Na-birnessite: enhanced cycling stability for sodium-ion batteries. CrystEngComm 2016;18:3136-41.
122. Liu Y, Chen Y, Wang J, et al. Hierarchical yolk-shell structured Li-rich cathode boosting cycling and voltage stabled LIBs. Nano Res 2021; doi: 10.1007/s12274-021-3890-1.
123. Xu M, Fei L, Zhang W, et al. Tailoring anisotropic Li-ion transport tunnels on orthogonally arranged Li-rich layered oxide nanoplates toward high-performance Li-ion batteries. Nano Lett 2017;17:1670-7.
124. Yabuuchi N, Hara R, Kajiyama M, et al. New O2/P2-type Li-excess layered manganese oxides as promising multi-functional electrode materials for rechargeable Li/Na batteries. Adv Energy Mater 2014;4:1301453.
125. Heubner C, Matthey B, Lein T, et al. Insights into the electrochemical Li/Na-exchange in layered LiCoO2 cathode material. Energy Storage Mater 2020;27:377-86.
126. Shang H, Zuo Y, Shen F, et al. O2-type Li0.78[Li0.24Mn0.76]O2 nanowires for high-performance lithium-ion battery cathode. Nano Lett 2020;20:5779-85.
127. Yang Z, Zhong J, Feng J, Li J, Kang F. Highly reversible anion redox of manganese-based cathode material realized by electrochemical ion exchange for lithium-ion batteries. Adv Funct Materials 2021;31:2103594.
128. Paulsen JM, Thomas CL, Dahn JR. Layered Li-Mn-oxide with the O2 structure: a cathode material for Li-ion cells which does not convert to spinel. J Electrochem Soc 1999;146:3560-5.
129. Robertson AD, Armstrong AR, Bruce PG. Layered LixMn1-yCoyO2 intercalation electrodes influence of ion exchange on capacity and structure upon cycling. Chem Mater 2001;13:2380-6.
130. Liu S, Zhu H, Zhang B, et al. Tuning the kinetics of zinc-ion insertion/extraction in V2O5 by in situ polyaniline intercalation enables improved aqueous zinc-ion storage performance. Adv Mater 2020;32:e2001113.
131. Yi T, Han X, Yang S, Zhu Y. Enhanced electrochemical performance of Li-rich low-Co Li1.2Mn0.56Ni0.16Co0.08-xAlxO2 (0≤x≤0.08) as cathode materials. Sci China Mater 2016;59:618-28.
132. Zhu J, Xiao G, Li X. Synthesis of Li(Ni0.6Co0.2Mn0.2)O2 by a modified sol-gel method for lithium-ion batteries. Synthetic Metals 2021;281:116905.
133. Xu G, Li J, Li X, et al. Understanding the electrochemical superiority of 0.6Li[Li1/3Mn2/3]O2-0.4Li[Ni1/3Co1/3Mn1/3]O2 nanofibers as cathode material for lithium ion batteries. Electrochimica Acta 2015;173:672-9.
134. Yu R, Zhang Z, Jamil S, et al. Effects of nanofiber architecture and antimony doping on the performance of lithium-rich layered oxides: enhancing lithium diffusivity and lattice oxygen stability. ACS Appl Mater Interfaces 2018;10:16561-71.
135. Zhou H, Ding X, Liu G, Gao Z, Xu G, Wang X. Characterization of cathode from LiNixMn2-xO4 nanofibers by electrospinning for Li-ion batteries. RSC Adv 2015;5:108007-14.
136. Ding X, Zhou H, Liu G, Yin Z, Jiang Y, Wang X. Electrochemical evaluation of LiAl0.05Ni0.05Mn1.9O4 cathode material synthesized via electrospinning method. J Alloys Compd 2015;632:147-51.
137. Zhou H, Ding X, Liu G, Jiang Y, Yin Z, Wang X. Preparation and characterization of ultralong spinel lithium manganese oxide nanofiber cathode via electrospinning method. Electrochimica Acta 2015;152:274-9.
138. Xu G, Li J, Li X, et al. The formation and electrochemical property of lithium-excess cathode material Li1.2Ni0.13Co0.13Mn0.54O2 with petal-like nanoplate microstructure. Ionics 2017;23:2285-91.
139. Liu H, Lei W, Tong Z, et al. Enhanced diffusion kinetics of Li ions in double-shell hollow carbon fibers. ACS Appl Mater Interfaces 2021;13:24604-14.
140. Zhou X, Wang F, Zhu Y, Liu Z. Graphene modified LiFePO4 cathode materials for high power lithium ion batteries. J Mater Chem 2011;21:3353.
141. Watanabe A, Matsumoto F, Fukunishi M, et al. Relationship between electrochemical pre-treatment and cycle performance of a Li-rich solid-solution layered Li1-α[Ni0.18Li0.20+αCo0.03Mn0.58]O2 cathode for Li-ion secondary batteries. Electrochemistry 2012;80:561-5.
142. Ito A, Li D, Ohsawa Y, Sato Y. A new approach to improve the high-voltage cyclic performance of Li-rich layered cathode material by electrochemical pre-treatment. J Power Sources 2008;183:344-6.
143. Ito A, Li D, Sato Y, et al. Cyclic deterioration and its improvement for Li-rich layered cathode material Li[Ni0.17Li0.2Co0.07Mn0.56]O2. J Power Sources 2010;195:567-73.
144. Wei Z, Xia Y, Qiu B, Zhang Q, Han S, Liu Z. Correlation between transition metal ion migration and the voltage ranges of electrochemical process for lithium-rich manganese-based material. J Power Sources 2015;281:7-10.
145. Yang J, Xiao L, He W, et al. Understanding voltage decay in lithium-rich manganese-based layered cathode materials by limiting cutoff voltage. ACS Appl Mater Interfaces 2016;8:18867-77.
146. Nayak PK, Grinblat J, Levi E, Markovsky B, Aurbach D. Effect of cycling conditions on the electrochemical performance of high capacity Li and Mn-rich cathodes for Li-ion batteries. J Power Sources 2016;318:9-17.
147. Pradon A, Caldes M, Petit P, et al. Revisiting the relevance of using a constant voltage step to improve electrochemical performances of Li-rich lamellar oxides. J Power Sources 2018;380:158-63.
148. Yin W, Grimaud A, Rousse G, et al. Structural evolution at the oxidative and reductive limits in the first electrochemical cycle of Li1.2Ni0.13Mn0.54Co0.13O2. Nat Commun 2020;11:1252.
149. Kaewmala S, Limphirat W, Yordsri V, et al. Rate dependent structural changes, cycling stability, and Li-ion diffusivity in a layered-layered oxide cathode material after prolonged cycling. J Mater Chem A 2021;9:14004-12.
150. Wang E, Zhao Y, Xiao D, et al. Composite nanostructure construction on the grain surface of Li-rich layered oxides. Adv Mater 2020;32:e1906070.
151. Yang K, Liu Y, Niu B, Yang Z, Li J. Oxygen vacancies in CeO2 surface coating to improve the activation of layered Li1.2Mn0.54Ni0.13Co0.13O2 cathode material for Li-ion batteries. Ionics 2019;25:2027-34.
152. Li J, Li J, Yu T, et al. Stabilizing the structure and suppressing the voltage decay of Li[Li0.2Mn0.54Co0.13Ni0.13]O2 cathode materials for Li-ion batteries via multifunctional Pr oxide surface modification. Ceram Int 2016;42:18620-30.
153. Ding F, Li J, Deng F, et al. Surface heterostructure induced by PrPO4 modification in Li1.2[Mn0.54Ni0.13Co0.13]O2 cathode material for high-performance lithium-ion batteries with mitigating voltage decay. ACS Appl Mater Interfaces 2017;9:27936-45.
154. Lu C, Wu H, Zhang Y, et al. Cerium fluoride coated layered oxide Li1.2Mn0.54Ni0.13Co0.13O2 as cathode materials with improved electrochemical performance for lithium ion batteries. J Power Sources 2014;267:682-91.
155. Lu C, Wu H, Chen B, Liu H, Zhang Y. Improving the electrochemical properties of Li1.2Mn0.52Co0.08Ni0.2O2 cathode material by uniform surface nanocoating with samarium fluoride through depositional-hydrothermal route. J Alloys Compd 2015;634:75-82.
156. Xue Q, Li J, Xu G, Zhou H, Wang X, Kang F. In situ polyaniline modified cathode material Li[Li0.2Mn0.54Ni0.13Co0.13]O2 with high rate capacity for lithium ion batteries. J Mater Chem A 2014;2:18613-23.
157. Chen Y, Wang X, Zhang J, et al. Al2O3-coated Li1.2Mn0.54Ni0.13Co0.13O2 nanotubes as cathode materials for high-performance lithium-ion batteries. RSC Adv 2019;9:2172-9.
158. Chen C, Geng T, Du C, et al. Oxygen vacancies in SnO2 surface coating to enhance the activation of layered Li-rich Li1.2Mn0.54Ni0.13Co0.13O2 cathode material for Li-ion batteries. J Power Sources 2016;331:91-9.
159. Liu Y, Yang Z, Li J, Niu B, Yang K, Kang F. A novel surface-heterostructured Li1.2Mn0.54Ni0.13Co0.13O2@Ce0.8Sn0.2O2-σ cathode material for Li-ion batteries with improved initial irreversible capacity loss. J Mater Chem A 2018;6:13883-93.
160. Yang Z, Zhong J, Li J, Liu Y, Niu B, Kang F. Li-rich layered oxide coated by nanoscale MoOx film with oxygen vacancies and lower oxidation state as a high-performance cathode material. Ceram Int 2019;45:439-48.
161. Liu Y, Yang Z, Zhong J, et al. Surface-functionalized coating for lithium-rich cathode material to achieve ultra-high rate and excellent cycle performance. ACS Nano 2019;13:11891-900.
162. Si M, Wang D, Zhao R, et al. Local electric-field-driven fast Li diffusion kinetics at the piezoelectric LiTaO3 modified Li-rich cathode-electrolyte interphase. Adv Sci (Weinh) 2020;7:1902538.
163. Zhang W, Sun Y, Deng H, et al. Dielectric polarization in inverse spinel-structured Mg2TiO4 coating to suppress oxygen evolution of Li-rich cathode materials. Adv Mater 2020;32:e2000496.
164. Hu S, Li Y, Chen Y, et al. Insight of a phase compatible surface coating for long-durable Li-rich layered oxide cathode. Adv Energy Mater 2019;9:1901795.
165. Cao J, Xie H, Lv F, et al. Stable Li2TiO3 Shell-Li1.17Mn0.50Ni0.16Co0.17O2 core architecture based on an in-site synchronous lithiation method as a high rate performance and long cycling life lithium-ion battery cathode. ACS Appl Energy Mater 2020;3:5462-71.
166. Yoon CS, Kim U, Park G, et al. Self-passivation of a LiNiO2 cathode for a lithium-ion battery through Zr doping. ACS Energy Lett 2018;3:1634-9.
167. Kong D, Hu J, Chen Z, et al. Ti‐gradient doping to stabilize layered surface structure for high performance high-Ni oxide cathode of Li-ion battery. Adv Energy Mater 2019;9:1901756.
168. Shang H, Ning F, Li B, Zuo Y, Lu S, Xia D. Suppressing voltage decay of a lithium-rich cathode material by surface enrichment with atomic ruthenium. ACS Appl Mater Interfaces 2018;10:21349-55.
169. Lee Y, Shin J, Kang H, et al. Promoting the reversible oxygen redox reaction of Li-excess layered cathode materials with surface vanadium cation doping. Adv Sci (Weinh) 2021;8:2003013.
170. Liu S, Liu Z, Shen X, et al. Surface doping to enhance structural integrity and performance of Li-rich layered oxide. Adv Energy Mater 2018;8:1802105.
171. Zhong J, Yang Z, Yu Y, Liu Y, Li J, Kang F. Surface substitution of polyanion to improve structure stability and electrochemical properties of lithium-rich layered cathode oxides. Appl Surf Sci 2020;512:145741.
172. Xu G, Li J, Xue Q, et al. Enhanced oxygen reducibility of 0.5Li2MnO3·0.5LiNi1/3Co1/3Mn1/3O2 cathode material with mild acid treatment. J Power Sources 2014;248:894-9.
173. Qiu B, Zhang M, Wu L, et al. Gas-solid interfacial modification of oxygen activity in layered oxide cathodes for lithium-ion batteries. Nat Commun 2016;7:12108.
174. Ye Z, Zhang B, Chen T, et al. A simple gas-solid treatment for surface modification of Li-rich oxides cathodes. Angew Chem Int Ed Engl 2021;60:23248-55.
175. Zhang X, Cao S, Yu R, et al. Improving electrochemical performances of Li-rich layered Mn-based oxide cathodes through K2Cr2O7 solution treatment. ACS Appl Energy Mater 2019;2:1563-71.
176. Zhang XD, Shi JL, Liang JY, et al. Suppressing surface lattice oxygen release of Li-rich cathode materials via heterostructured spinel Li4Mn5O12 coating. Adv Mater 2018:e1801751.
177. Hu K, Lv G, Zhang J, et al. Na2S treatment and coherent interface modification of the Li-rich cathode to address capacity and voltage decay. ACS Appl Mater Interfaces 2020;12:42660-8.
178. Zhang K, Sheng H, Wu X, et al. Improving electrochemical properties by sodium doping for lithium-rich layered oxides. ACS Appl Energy Mater 2020;3:8953-9.
179. Zhu Z, Wang H, Li Y, et al. A surface Se-substituted LiCo[O2-δSeδ] cathode with ultrastable high-voltage cycling in pouch full-cells. Adv Mater 2020;32:e2005182.
180. Ahn J, Chen D, Chen G. A fluorination method for improving cation-disordered rocksalt cathode performance. Adv Energy Mater 2020;10:2001671.
181. Liu C, Wu M, Guo Z, et al. Preparation and characterization of Li1.167-xKxMn0.583Ni0.25O2 (x=0, 0.025, 0.05 and 0.075) as cathode materials for highly reversible lithium-ion batteries. Electrochimica Acta 2020;341:136014.
182. Li B, Yan H, Ma J, et al. Manipulating the electronic structure of Li-rich manganese-based oxide using polyanions: towards better electrochemical performance. Adv Funct Mater 2014;24:5112-8.
183. Wu F, Kim G, Kuenzel M, et al. Elucidating the effect of iron doping on the electrochemical performance of cobalt-free lithium-rich layered cathode materials. Adv Energy Mater 2019;9:1902445.
184. Nayak PK, Grinblat J, Levi M, et al. Al doping for mitigating the capacity fading and voltage decay of layered Li and Mn-rich cathodes for Li-ion batteries. Adv Energy Mater 2016;6:1502398.
185. Jiang Y, Sun G, Yu F, et al. Surface modification by fluorine doping to increase discharge capacity of Li1.2Ni0.2Mn0.6O2 cathode materials. Ionics 2020;26:151-61.
186. Li Y, Xie L, Zheng Z, et al. Hybridizing Li@Mn6 and Sb@Ni6 superstructure units to tune the electrochemical performance of Li-rich layered oxides. Nano Energy 2020;77:105157.
187. Wang T, Zhang C, Li S, et al. Regulating anion redox and cation migration to enhance the structural stability of Li-rich layered oxides. ACS Appl Mater Interfaces 2021;13:12159-68.
188. Yu F, Que L, Xu C, et al. Dual conductive surface engineering of Li-Rich oxides cathode for superior high-energy-density Li-Ion batteries. Nano Energy 2019;59:527-36.
189. Luo S, Wang K, Wang J, Jiang K, Li Q, Fan S. Binder-free LiCoO2/carbon nanotube cathodes for high-performance lithium ion batteries. Adv Mater 2012;24:2294-8.
190. Zhang T, Li JT, Liu J, et al. Suppressing the voltage-fading of layered lithium-rich cathode materials via an aqueous binder for Li-ion batteries. Chem Commun (Camb) 2016;52:4683-6.
191. Yang J, Li P, Zhong F, et al. Suppressing voltage fading of Li-rich oxide cathode via building a well-protected and partially-protonated surface by polyacrylic acid binder for cycle-stable Li-ion batteries. Adv Energy Mater 2020;10:1904264.
192. Choi Y, Zhang K, Chung KY, Wang DH, Park JH. PVdF-HFP/exfoliated graphene oxide nanosheet hybrid separators for thermally stable Li-ion batteries. RSC Adv 2016;6:80706-11.
193. Shin WK, Kannan AG, Kim DW. Effective suppression of dendritic lithium growth using an ultrathin coating of nitrogen and sulfur codoped graphene nanosheets on polymer separator for lithium metal batteries. ACS Appl Mater Interfaces 2015;7:23700-7.
194. Zhang J, Meng Z, Yang D, et al. Enhanced interfacial compatibility of FeS@N,S-C anode with ester-based electrolyte enables stable sodium-ion full cells. J Energy Chem 2022;68:27-34.
195. Qi S, Mi L, Song K, et al. Understanding shuttling effect in sodium ion batteries for the solution of capacity fading: FeS2 as an example. J Phys Chem C 2019;123:2775-82.
196. Martha SK, Nanda J, Veith GM, Dudney NJ. Surface studies of high voltage lithium rich composition: Li1.2Mn0.525Ni0.175Co0.1O2. J Power Sources 2012;216:179-86.
197. Cui C, Fan X, Zhou X, et al. Structure and interface design enable stable Li-rich cathode. J Am Chem Soc 2020;142:8918-27.
198. Lou S, Ma Y, Zhou Z, et al. Unravelling the enhanced high-temperature performance of lithium-rich oxide cathode with methyl diphenylphosphinite as electrolyte additive. ChemElectroChem 2018;5:1569-75.
199. Yue Y, Li N, Li L, et al. Redox behaviors in a Li-excess cation-disordered Mn-Nb-O-F rocksalt cathode. Chem Mater 2020;32:4490-8.
200. Yu T, Li J, Xu G, Li J, Ding F, Kang F. Improved cycle performance of Li[Li0.2Mn0.54Co0.13Ni0.13]O2 by Ga doping for lithium ion battery cathode material. Solid State Ionics 2017;301:64-71.
201. Redel K, Kulka A, Plewa A, Molenda J. High-performance Li-rich layered transition metal oxide cathode materials for Li-ion batteries. J Electrochem Soc 2019;166:A5333-42.
202. Hy S, Felix F, Rick J, Su WN, Hwang BJ. Direct in situ observation of Li2O evolution on Li-rich high-capacity cathode material, Li[Ni(x)Li((1-2x)/3)Mn((2-x)/3)]O2 (0 ≤ x ≤ 0.5). J Am Chem Soc 2014;136:999-1007.
203. Sathiya M, Leriche JB, Salager E, Gourier D, Tarascon JM, Vezin H. Electron paramagnetic resonance imaging for real-time monitoring of Li-ion batteries. Nat Commun 2015;6:6276.
204. Xu J, Sun M, Qiao R, et al. Elucidating anionic oxygen activity in lithium-rich layered oxides. Nat Commun 2018;9:947.
205. Zhang J, Wang Q, Li S, et al. Depth-dependent valence stratification driven by oxygen redox in lithium-rich layered oxide. Nat Commun 2020;11:6342.
206. Dai K, Wu J, Zhuo Z, et al. High reversibility of lattice oxygen redox quantified by direct bulk probes of both anionic and cationic redox reactions. Joule 2019;3:518-41.
207. Shimoda K, Minato T, Nakanishi K, et al. Oxidation behaviour of lattice oxygen in Li-rich manganese-based layered oxide studied by hard X-ray photoelectron spectroscopy. J Mater Chem A 2016;4:5909-16.
208. Assat G, Iadecola A, Foix D, Dedryvère R, Tarascon J. Direct quantification of anionic redox over long cycling of Li-rich NMC via hard X-ray photoemission spectroscopy. ACS Energy Lett 2018;3:2721-8.
209. Abate II, Pemmaraju CD, Kim SY, et al. Coulombically-stabilized oxygen hole polarons enable fully reversible oxygen redox. Energy Environ Sci 2021;14:4858-67.
210. House RA, Maitra U, Pérez-Osorio MA, et al. Superstructure control of first-cycle voltage hysteresis in oxygen-redox cathodes. Nature 2020;577:502-8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Yang Z, Zheng C, Wei Z, Zhong J, Liu H, Feng J, Li J, Kang F. Multi-dimensional correlation of layered Li-rich Mn-based cathode materials. Energy Mater 2022;2:200006. http://dx.doi.org/10.20517/energymater.2022.02
AMA Style
Yang Z, Zheng C, Wei Z, Zhong J, Liu H, Feng J, Li J, Kang F. Multi-dimensional correlation of layered Li-rich Mn-based cathode materials. Energy Materials. 2022; 2(1): 200006. http://dx.doi.org/10.20517/energymater.2022.02
Chicago/Turabian Style
Yang, Zhe, Chaoliang Zheng, Zhicheng Wei, Jianjian Zhong, Huirong Liu, Jiameng Feng, Jianling Li, Feiyu Kang. 2022. "Multi-dimensional correlation of layered Li-rich Mn-based cathode materials" Energy Materials. 2, no.1: 200006. http://dx.doi.org/10.20517/energymater.2022.02
ACS Style
Yang, Z.; Zheng C.; Wei Z.; Zhong J.; Liu H.; Feng J.; Li J.; Kang F. Multi-dimensional correlation of layered Li-rich Mn-based cathode materials. Energy Mater. 2022, 2, 200006. http://dx.doi.org/10.20517/energymater.2022.02
About This Article
Copyright

Data & Comments
Data


 Cite This Article 25 clicks
Cite This Article 25 clicks

 Like This Article 25
likes
Like This Article 25
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.