
Accelerating perovskite materials discovery and correlated energy applications through artificial intelligence

Abstract
Perovskites are promising materials applied in new energy devices, from solar cells to battery electrodes. Under traditional experimental conditions in laboratories, the performance improvement of new energy devices is slow and limited. Artificial intelligence (AI) has recently drawn much attention in material properties prediction and new functional materials exploration. With the advent of the AI era, the methods of studying perovskites have been upgraded, thereby benefiting the energy industry. In this review, we summarize the application of AI in perovskite discovery and synthesis and its positive influence on new energy research. First, we list the advantages of AI in perovskite research and the steps of AI application in perovskite discovery, including data availability, the selection of training algorithms, and the interpretation of results. Second, we introduce a new synthesis method with high efficiency in cloud labs and explain how this platform can assist perovskite discovery. We review the use of perovskites in energy applications and illustrate that the efficiency of energy production in these fields can be significantly boosted due to the use of AI in the development process. This review aims to provide the future application prospects of AI in perovskite research and new energy generation.
Keywords
INTRODUCTION
As a result of increasing environmental issues, the energy crisis is now a severe problem for humans globally due to climate change and fossil fuel depletion[1,2]. Besides reducing fuel consumption and carbon-neutral policies, topics regarding advanced energy materials for efficient energy generation, consumption, and storage have attracted significant attention, including batteries[3-8], photocatalysts[9-11], supercapacitors[12-14], solar cells[15,16] and fuel cells[17-19]. Batteries and cells can further be improved by electrolyte, cathode and anode materials discovery[20-22]. However, the performance of current energy materials is not satisfactory for the ongoing energy crisis.
Recently, the types of energy materials are increasing rapidly and more attention is being paid to the potential of perovskites in the energy sector. Perovskite materials (PMs) are becoming popular because of their novel optoelectronic properties. PMs have a general formula of ABX3, where A is an inorganic ion or organic cation, B is a metal ion, and X is a halide anion. This flexible structure typically results in an excellent variety of PMs with different properties in the organic and inorganic regions. PMs now have wide applications in photocatalysts[23], perovskite solar cells (PSCs)[24-26] and batteries[27]. Figure 1A shows the general PM structure and the structure of a highly stable, hole-conductor-free, and printable mesoscopic PSC[25]. Due to the diversity of PMs and energy device architectures, PSCs can be constructed with different PMs with various performances, stabilities, and costs, as shown in Figure 1B[26]. However, the traditional method of PM research is a time-consuming process. To improve the time cost in the discovery and performance of PM applications, researchers now devote more attention to the assistance of artificial intelligence (AI).
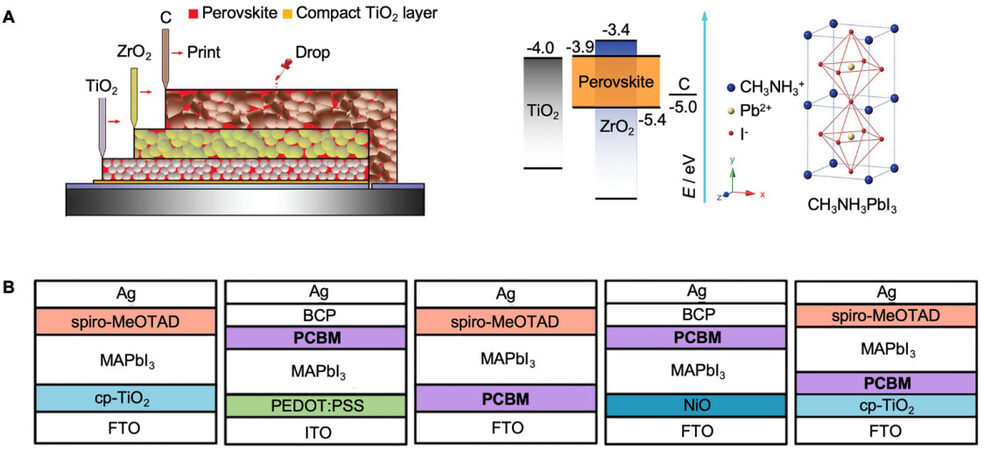
Figure 1. Recent energy materials, perovskite structure description, and energy material application. (A) Perovskite material structure and architecture of a highly stable, hole-conductor-free, and printable mesoscopic perovskite solar cell. Copyright from AAAS[25]. (B) Different compositions of perovskite solar cells showing the variety of the perovskite family and its potential. Copyright from ACS Publications[26].
AI is not a novel technology, having been developed rapidly in recent decades and applied to chemistry and materials science in seeking new structures with higher performance. AI technology helps us to discover the correlation between properties and structures. AI technology has several branches, including machine learning (ML) and artificial neural networks (ANNs). ML uses models and algorithms to understand high-dimensional features in the supplied data. Choudhary et al.[28] used ML methods to explore efficient solar cell PMs by combining them with density functional theory (DFT) calculations through the Vienna Ab Initio Simulation Package (VASP). DFT software, like VASP[28], also helps to generate training data. Therefore, AI abilities offer a possible solution in performing various PM discoveries and applications.
This review presents an in-depth assessment of AI technology assistance for PMs and how AI can help to improve PM performance in the energy region, especially for solar cells and batteries. The following section starts from the AI workflow, including database information, model introduction, and interpretable ML, which help researchers comprehend ML. After training and predictions, we introduce AI-assisted synthesis and the concept of cloud labs, which accelerate new PM generation and testing. After synthesis, the comparison is made between the performance of general and AI-assisted PM productions. Energy conversion efficiency and capacity are reviewed. In the final section, we give our perspectives on how AI can assist PM discovery in other sectors and future challenges.
OBJECTIVES OF AI AND ITS WORKFLOW
AI for energy materials
PMs are favorable candidates for the next generation of energy devices. Given the high chemical flexibility made available by the perovskite framework in accommodating a broad spectrum of atomic substitutions and the multiple possibilities of spanning compositions or configurations of double perovskites, double perovskite oxides are considered to be promising materials that are beneficial for energy conversion and storage, e.g., PSCs[29]. With the commercialization of PMs, new perovskite-related performance requirements challenge related research efforts. For instance, the stability of PMs is limited by restricting photovoltaic device lifetimes to 3000 h[30]. In addition, other topics, such as predicting the formability of perovskite structures of hybrid organic-inorganic perovskites (HOIPs), also pose challenges for the space exploration of PMs[7]. However, traditional trial-and-error approaches to perovskite-related research and development, e.g., PSC screening and stability testing, are labor-intensive and expensive. The high computational cost results from the high-dimensional perovskite parameter space, multiple environmental factors (light, temperature, bias, oxygen, and humidity), many possible compound compositions enumerated, and the calculation of physical properties, such as high-throughput DFT, the GW approach, and hybrid functionals for bandgap estimation[31,32].
Research removes the burden of traversing every possible combination and accelerates progress with data-driven approaches and has recently become a remarkable route. There have already been massive open-access databases of computing materials properties, recording information on electronic structure, thermodynamic and structural properties. It is possible to find efficient ways to extract knowledge for materials science with databases. Therefore, ML is gradually making inroads into materials science, where one can predict the properties of materials with features efficiently yet accurately. AI allows machines to develop knowledge and perform human-like tasks, such as materials science research. The brain of AI is ML, an interdisciplinary subject that includes computer science, statistics, and mathematics. The goal of ML is to construct a model under the guidance of an algorithm to develop knowledge from historical data, and thus it can evaluate or predict new objects.
ML is an ideal toolkit to accelerate PM research and development at an unprecedented pace. Over the last decade, ML has been applied to materials science problems in a variety of directions for properties prediction, such as the formation energy of elpasolite structures, molecular electronic properties in chemical compound space, the density of electronic states at the Fermi energy, the molecular atomization energies of molecules, the Curie temperature of high-temperature piezoelectric perovskites, the thermodynamic stability of ternary oxide compounds, the bandgap energy (Eg) of crystalline compounds and the metallic glass-forming ability of ternary amorphous alloys, crystal structures and the development of interatomic potentials[33]. Furthermore, ML can find the optimal density functionals for DFT and build predictive models of material properties[33]. Moreover, ML has applications in related fields, such as energy storage, where various research groups have implemented models to forecast the remaining lifetime of batteries and fuel cells[31]. ML models also can predict underlying physical phenomena, as well as PSC performance. Even though it is nearly impossible for researchers to find relevant patterns from a dataset, the PSC model predictions closely match the theoretical predictions of the Shockley and Queisser limits. Instead of the previous computational materials design, which derived materials properties according to physical laws, ML can obtain latent structural or compositional information from the big data and eliminate the practical obstacles for synthesizing PMs. The general workflow of ML in material science, shown in Figure 2[34], includes data preparation, feature engineering and model selection, evaluation, and application. The model applications can guide the process of target PMs.
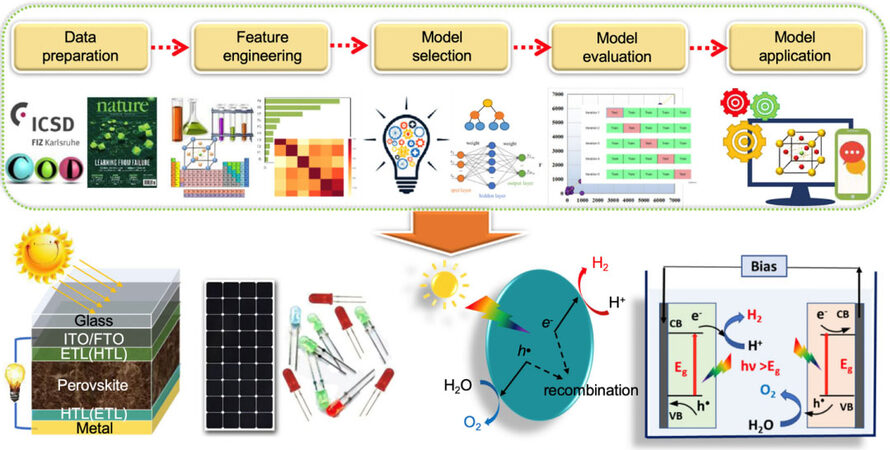
Figure 2. AI accelerating perovskite structure discovery with relevant data. The figure shows the general workflow of machine learning in materials science. Copyright from Springer Nature[34].
Available databases and data preparation
Open-access databases of material properties provide a solid foundation for ML applications. Since the data determine the upper bound of ML performance, it is significant to use high-quality data to prevent erroneous and redundant information for ML. Table 1 provides a list of authoritative open databases containing information regarding perovskites. The commonly used authoritative open databases are the Materials Project[35], the Open Quantum Materials Database (OQMD)[36], and the Computational Materials Repository (CMR)[37]. The Materials Project, developed by Lawrence Berkeley National Laboratory (Berkeley Lab) and the Massachusetts Institute of Technology (MIT), uses supercomputing and state-of-the-art electronic structure methods to uncover the properties of all known inorganic materials. The latest database release V2021.03.13 features a new formation energy correction scheme. The OQMD is a high-throughput database currently consisting of nearly 300,000 DFT total energy calculations of compounds from the Inorganic Crystal Structure Database (ICSD)[38] and illustrations of commonly occurring crystal structures. It contains 3486 perovskites with symmetrically equivalent sites and 6972 perovskites with symmetrically distinct sites. The ICSD is the world’s largest database of entirely determined inorganic crystal structures, from elements to quintenary compounds. It contains about 185,000 structures, with 6,000 added annually. Each record includes crystallographic data, chemical/physical property data, and bibliographic information referencing the journal article structure. The CMR addresses the data challenge of quantum physical calculations and provides a software infrastructure that supports the collection, storage, retrieval, analysis, and sharing of data produced by many electronic structure simulators. The records were obtained by combining 53 stable cubic perovskite oxides with a finite bandgap on single perovskites[32]. These 53 parent single perovskites contained fourteen different A-site cations and ten B-site cations.
Publicly accessible databases containing perovskite-related data
| Database | Brief Description | URL |
| Materials Project | It uses high-throughput computing to uncover the properties of all known inorganic materials. | http://materialsproject.org/ |
| The Open Quantum Materials Database (OQMD) | A high-throughput database currently consists of nearly 300,000 DFT total energy calculations of compounds from the ICSD and decorations of commonly occurring crystal structures. | http://oqmd.org |
| Computational Materials Repository (CMR) | The Computational Materials Repository addresses this data challenge of quantum physics calculations. It provides a software infrastructure that supports the collection, storage, retrieval, analysis, and sharing of data produced by many electronic-structure simulators. | https://cmr.fysik.dtu.dk/ |
| AFLOW | An automatic software framework for high-throughput materials discovery. | http://www.aflowlib.org/ |
| Inorganic Crystal Structure Database (ICSD) | It is the world's largest database of fully determined inorganic crystal structures, from elements to quintenary compounds. Each record includes crystallographic data, chemical/physical property data, and bibliographic information referencing the journal article structure. | https://icsd.products.fiz-karlsruhe.de/ |
| Cambridge Structural Database (CSD) | The Cambridge Structural Database (CSD) contains a complete record of all published organic and metal-organic small-molecule crystal structures. Before entering the database, all structures are processed computationally and by expert structural chemistry editors. A key component of this processing is the reliable association of the chemical identity of the structure studied with the experimental data. This important step helps ensure that data is widely discoverable and readily reusable. | https://scripts.iucr.org/cgi-bin/paper?bm5086 |
| JARVIS | JARVIS (Joint Automated Repository for Various Integrated Simulations) is a repository designed to automate materials discovery and optimization using classical force-field, DFT, ML calculations, and experiments. | https://jarvis.nist.gov/ |
| Crystallography Open Database (COD) | COD collects all known small molecule or small to medium-sized unit cell crystal structures and makes them available freely on the Internet. As of today, the COD has aggregated ~150,000 structures, offering basic search capabilities and the possibility to download the whole database, or parts thereof using a variety of standard open communication protocol | http://www.crystallography.net |
| Springer Materials | The platform provides the most comprehensive and multidisciplinary collection of materials and chemical properties with extensive coverage of all major topics in materials science and related disciplines, taking advantage of the best and most trusted materials science sources such as Landolt Börnstein data on a single platform. | https://materials.springer.com/ |
Many datasets contain PMs for energy applications that scientists have collected from works in recent decades[36-42]. Recent work has also discussed data augmentation strategies. For example, Oviedo et al.[43] performed peak scaling, peak elimination, and pattern shifting to augment an XRD dataset based on physics domain knowledge. Xu et al.[44] claimed that, based on the currently known derived data on the formability of perovskites with 2,000 compositions under certain environmental pressures, the number of stable perovskites is expected to reach 90,000.
Perovskite modeling and training procedure
Feature engineering
Besides the raw data, another important factor determining the effectiveness of ML models is how we describe the properties. The description should be physically meaningful, chemically intuitive, and consistent with materials transformations[32]. In most cases, the relationship between the primary features and the target is unlikely to be linear. With the primary features, conjunctive features are formed to allow for nonlinearity in the linear models. Normalization is another important operation to adjust feature distribution to the standard normal distribution, ensuring they are on the same scale. Some ML models are sensitive to feature scalings, such as the neural network (NN) and the support vector machine (SVM).
Besides those listed above, dimension reduction is yet another determinative operation in high-dimensional feature spaces and chemical data are typically high dimensional. High-dimensional features lead to high computational complexity, the curse of dimensionality, and the disappearance of information due to multicollinearity. There are two general methods for dimension reduction: feature selection and linear transformation. For many classical ML models, feature selection is a key factor in determining a successful model since it reduces the complexity of the model space, helps avoid overfitting and eliminate unrelated features and noise. Furthermore, it can also shorten the training time and further promote the prediction ability and generalization performance of the model. An intuitive method to perform feature selection is to drop the features with a high Pearson correlation. Recent works also propose an algorithm-based method to select the features, e.g., LASSO and genetic and greedy algorithms[45]. The linear transformation for dimension reduction is often achieved through matrix decomposition techniques, such as singular value decomposition and principal component analysis (PCA). PCA is the most popular method because it allows for transforming the parameter space into a mutually independent parameter space with a given dimension by selecting the first N eigenvalues of the covariance matrix of the parameter matrix. As a result, PCA eliminates complex computation problems, the curse of dimension, and multicollinearity. However, PCA may not provide the optimal principal component for non-Gaussian distribution data.
Model selection
ML algorithms can be grouped into supervised, unsupervised, and reinforcement learning. The choice of model mainly depends on the type of task. Supervised learning is the primary choice for a target output, such as the Eg of crystalline compounds. Supervised learning models can be further divided into regression and classification models, corresponding to continuous and discrete output items. If the main task is to infer or analyze data and is without any notation regarding relation, then the corresponding ML algorithm is unsupervised learning. Simultaneously, reinforcement learning suits the tasks rewarded by environment interaction. Deep learning is generally applied in supervised and reinforcement learning, but it requires significant data to perform well. In general, the best model is an ensemble algorithm, which is obtained by combining multiple algorithms. We display a flowchart in Figure 3[46] that can assist in rapid model selection. Cross-validation and independent testing are the primary basis for evaluating models. Commonly used evaluation indicators include the mean absolute error (MAE), mean square error (MSE), root mean square error (RMSE), coefficient of determination (R2), and regression correlation coefficient (R), with the confusion matrix, precision, recall, test receiver operating characteristic curve (ROC) and area under the ROC curve (AUC) were used for classification.
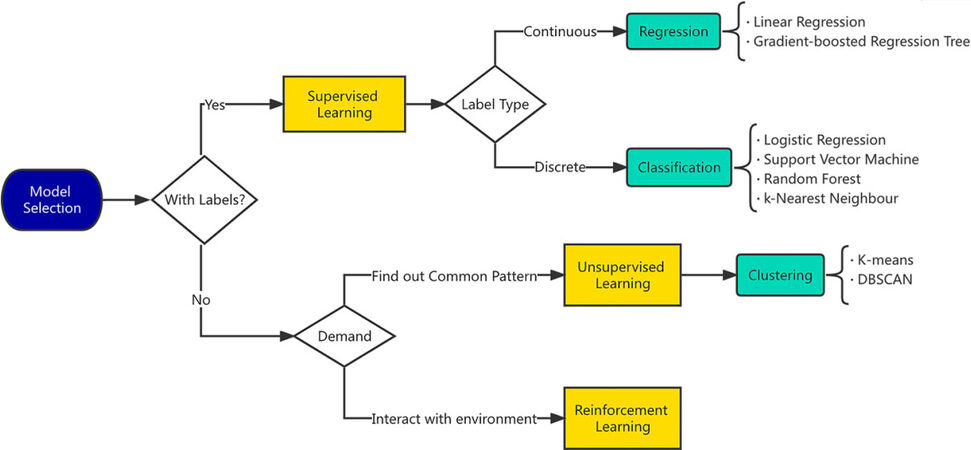
Figure 3. Decision flowchart of model selection in common ML tasks[46], which is applied uniformly across perovskite discovery and properties prediction. The DBSCAN on the right part means “density-based spatial clustering of applications with noise”.
The terms “machine learning” and “deep learning” have become very popular in recent years, but both are confusing. ML is a part of AI that focuses on imitating human learning, while deep learning is one of the research orientations of ML. ML includes many famous algorithms, such as linear regression, decision trees, Bayesian learning and ANNs. ML extends from statistics, information theory and matrix analysis to obtain the optimal solution rivaling human learning results. For instance, random forest (RF) is an ensemble learning method that uses multiple decision trees for classification and prediction, while each decision tree splits the node by maximum information gain. In addition, following the principal theory of the ML model, the model can explain the relationship between the features and the target. However, traditional ML is not sufficiently intelligent to handle complex problems, such as image recognition, speech recognition, and natural language processing, and deep learning is therefore proposed. Deep learning originates from the ANN. The ANN is an algorithm with nonlinear adaptive information processing capability, consisting of multiple hidden-layer perceptions, and is the basic framework of deep learning. Deep learning is intelligent because it incorporates a complex algorithm that learns and reorganizes lower-layer perceptions to form abstract but efficient, higher-layer neurons for the final decision. With this mechanism, deep learning is superior to previous technologies in complex problems. Complicated systems can also be problematic. Firstly, deep learning requires tremendous amounts of data, usually at the million level, to overcome underfitting if trained from scratch. Moreover, significant computing resources will be spent to train a model, resulting in substantial costs. In addition, deep learning models are complicated to interpret because of their complexity, which means they cannot indicate patterns in the data.
Model evaluation and validation
The core of supervised learning is to infer the unknown from the known. There will inevitably be some statistical errors in the algorithm operation, and model evaluation is required to ensure the validity of the ML model and the correctness of the results. Generalization ability is an important indicator for evaluating models, which refers to the adaptability of ML algorithms to fresh samples. The purpose of ML is to find the laws hidden behind the data. The trained model can also give appropriate output for data other than the training set with the same laws. Therefore, the key is using the test set to test the generalization ability. It is noteworthy that the test and training sets need to be mutually exclusive. The error obtained using the test set can be seen as an approximation of the generalization error.
A commonly used method for evaluating the reliability of ML models is k-fold cross-validation (k-fold CV). K-fold CV is to divide the input data into k mutually exclusive subsets of similar size based on stratified sampling. The union of k-1 subsets is then used as the training set and the remaining one is used as the test set. After k training and testing, the final ML performance is the average of all test results. The k value of the k-fold CV method, i.e., the number of subsets, has a significant impact on the stability and fidelity of the evaluation results. Commonly used values for k are 5, 10, and 20. When k is equal to the number of input data samples, k-fold CV becomes leave-one-out cross-validation (LOOCV). LOOCV is not affected by random sample division. The results are generally considered to be more accurate but simultaneously result in greater time costs and computational resource consumption. Whether it is a common test set or k-fold CV, quantitative evaluation metrics are required to measure model performance. Different algorithm tasks have different evaluation metrics. For regression algorithms, the commonly used evaluation metrics are MSE and R2. For classification algorithms, the commonly used metrics are precision, recall, precision, F1 score, ROC, and AUC.
Interpretable ML
Interpretable models
It is common to apply interpretable models in materials science because we want to know what property affects the final performance of an energy material. Through k-fold CV, the support vector regressor (SVR) model is a very efficient method for predicting the Curie temperature (Tc) of PMs, resulting in an R of 0.8549, an RMSE of 28.6659, and an MRE of 0.0725[45]. An explainable strategy is proposed that combines ML with the Shapley Additive Explanations (SHAP) method to accelerate the discovery of potential HOIPs[7]. The most common interpretable models in PM research are tree models, including RF[47] and gradient boosting regression tree (GBRT) models[33]. Im et al.[33] used a GBRT model to predict heats of formation and bandgaps, and a statistical analysis of the selected features identified design guidelines for discovering new lead-free perovskites. On the test set, the GBRT model was more accurate in predicting the heats of formation but had a more significant prediction error for the bandgaps. The importance scores of all features in the predictions of the heats of formation and bandgaps given by this GBRT model are shown in Figure 4A. As can be seen, in the GBRT prediction of the heats of formation, the halogen anion type (Xx) is the most crucial feature. The importance score of DB3+, the second important feature, has been reduced by half and the later feature importance score has become negligible. The distribution of important scores implies that the heat of formation strongly depends on halide anions and DB3+. In contrast, when the GBRT predicts Eg, the most important feature SG score is almost insignificant, indicating a more complex relationship between Eg and material features.
Deep learning
Deep learning has also excelled in materials science research in recent years. The highly nonlinear nature of deep learning can more fully restore the underlying physical mechanism. Kirman et al.[47] published a high-throughput experimental framework in 2020 to discover new perovskite single crystals. This framework put high-throughput synthesized perovskite single-crystal images into a convolutional neural network (CNN) to obtain characterization results to predict the optimal conditions for synthesizing new perovskite single crystals and report the first synthesis of (3-PLA)2PbCl4. Saidi et al.[48] used a deep learning model to predict Eg ranging from 0.2 to 6.0 eV in the same year. The CNN performed exceptionally well, delivering a bandgap RMSE of only 0.02 eV compared to DFT results.
The above are examples of the further characterization or direct prediction of material properties with the help of deep learning. The question of whether deep learning models can explain physical and chemical laws is another direction of the discussion. Due to its high degree of nonlinearity and complexity, deep learning is considered a black box, reflecting its low interpretability. Nevertheless, some materials scientists have developed explainable deep learning models. A crystal graph convolutional neural network (CGCNN) was proposed in 2018 to represent periodic crystal systems that provide material property predictions and atomic-level chemical insights with DFT precision[46]. With multiple convolutional, pooling, and hidden layers, the CGCNN can extract any structural differences based on atomic connections and discover latent relationships between structures and properties. Simultaneously, the empirical rules derived from the model results are consistent with the obvious aim of finding more stable perovskites, implying a reduced search space for high-throughput screening. Figure 4B shows the site energy distribution obtained by the CGCNN on sites A and B, respectively. Assuming that the cutoff energy for potential synthesizable is set to 0.2 eV/atom, PbMoO3 falls within a reasonable range. It can be synthesized successfully, which confirms that the chemical insights gained from the CGCNN reduce the search space for high-throughput screening, thereby increasing the material search efficiency by a factor of seven.
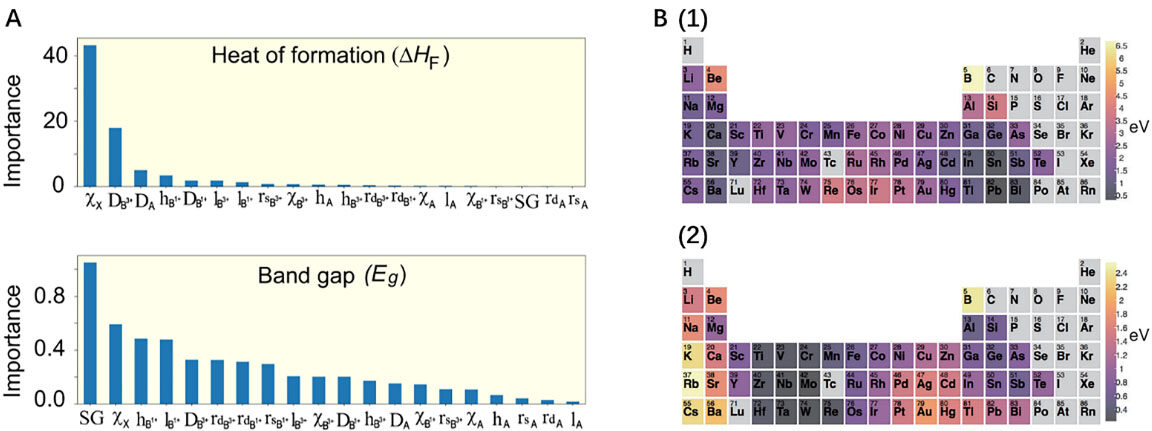
Figure 4. Interpretation of perovskite ML models. (A) GBRT model gives insight into the heat of formation and bandgap predictions. The result shows that Xx plays an important role in both the heat of formation and bandgap. Copyright from Springer Nature[33]. (B) Mean site energy when the element occupies site A (1) and site B (2) is calculated by the CGCNN model. Copyright from APS[46].
The balance between accuracy, the interpretability of predictive models, and theoretical consistency is an essential proposition in the use of ML in materials science. Due to the complex interactions between material components, the relationship between material features and target properties is usually highly nonlinear, requiring the fitting of flexible nonlinear ML algorithms. However, most nonlinear ML models lack interpretability, adding difficulty to mechanistic understanding, such as finding critical components of target properties. Therefore, finding a balance of accurate prediction and interpretability by ML algorithms is crucial to advance data-driven materials research further. In addition, the consistency between the model interpretation and the theories in physics and chemistry is noteworthy. The model will be valueless if its interpretation does not match the theories, leading to the unachievable synthesis of new PMs.
IMPROVING PEROVSKITE PERFORMANCE IN ENERGY APPLICATIONS
Performance evolution of perovskite applications
Since PMs have been widely used in energy devices[24,49], improving their performance is the next step. In 2013, the efficiency of a planar heterojunction solar cell with a CH3NH3PbI3-xClx absorber layer could reach an energy conversion efficiency of ~15%[50]. In 2021, He et al.[51] doped MAPbI3 into a homojunction PSC with NiO as the hole transport layer and the best efficiency reached 19.101%. Many new perovskite structures and devices are reported each year, and researchers are curious regarding the tendency of PM evolution in terms of composition and/or structure. Odabaşı et al.[52] reported that ML can screen perovskite structures and take part in an automatic synthesis system and help understand this tendency by learning the data extracted from previous works. One thousand nine hundred twenty-one records of (organo)-lead-halide PSC device performance were collected from 800 publications from 2013 to 2018. Figure 5A shows their collected samples sorted by publication year and efficiency. The average efficiency of all three cell structures increased from ~8% to ~14%. Other conditions, like deposition procedures, solvents, anti-solvents, electron transport layer materials, and hole transport layer materials, are all sorted by year and analyzed on cell efficiency, which have increased over the years.
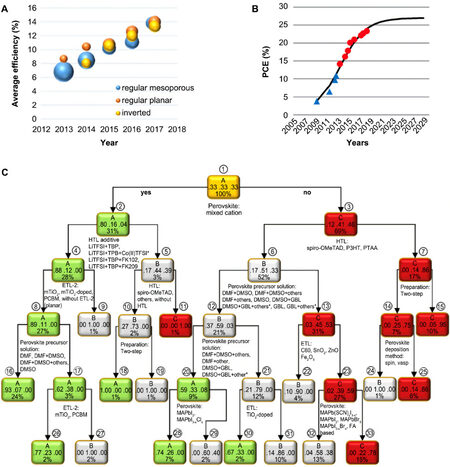
Figure 5. ML-assisted understanding of previous works on PSCs. (A) Discrete plot of average efficiency over the years legend by cell structure types, implying an increasing trend. (B) Forecast of cell efficiency growth by logistic growth model. (C) Decision tree of PSC efficiency based on synthesis conditions and PM material types. Copyright from Elsevier[52]. PCE: Power conversion efficiency.
Based on these data, a logistic growth model is generated to predict the efficiency limit of PSCs with blue points from Ye et al.[53] and red points from NREL, as shown in Figure 5B. They also predicted the stabilized efficiencies of normal cells using the decision tree in Figure 5C. The A, B, and C classes denote high efficiency (> 18%), intermediate efficiency, and low efficiency (< 9%), respectively. By obeying classification rules, the fraction of each node is present at the bottom of the frame. The middle of the frame shows the fraction of the A, B, and C classes in that node. The color and the top of the frame mean the class with the highest fraction in this node. The advantage of the decision tree is that it directly shows the interpretation of rules on the branches. Future improvements can be made by following the arrows on this diagram, but the limitation displayed in Figure 5B is a problem. Detailed predictions of properties are needed.
Improvement of desired properties
Rather than strictly following development laws, focusing on specific properties can distinctly improve the performance of a device. Different combinations of the A and B cations in the ABX3 structure result in different performances, and appropriate doping or a double perovskite can significantly improve the desired properties. This means that the form of doping a PM changes into (A1-yMy)a(B1-yNy)bXc, where a and b can be unequal. This form raises a new problem, namely, finding an optimal doping value that is nearly continuous in an extensive perovskite system. Sun et al.[54], as mentioned above, used a DNN to solve this problem with a high-throughput experiment system. They classified synthetic Cs3(Bi1-xSbx)2(I1-xBrx)9 compounds, shown in Figure 6A, into 0D, 2D, and 3D structures according to the XRD data in Figure 6B. Although they practically trained their DNN with limited data (164 PXRD data from the ICSD), the system showed a high accuracy of over 90% in classification. The experiment was ten times faster than human labor. The interpretation of the ML model also gave suggestions regarding structure types. When x is equal to 20%, the model shows a high confidence score, indicating that the PM is in a 2D phase structure [Figure 6C], and by increasing x, tighter binding and larger bandgaps are achieved [Figure 6E]. The absorptance information agrees with this result in Figure 6D. The interpretation shows that the bandgap trend is not dependent on direct or indirect bandgap trends, but perhaps on the x value. Determining the optimal x can further understand the bandgap bowing phenomenon, which is observed in several common semiconductors and solar cells.
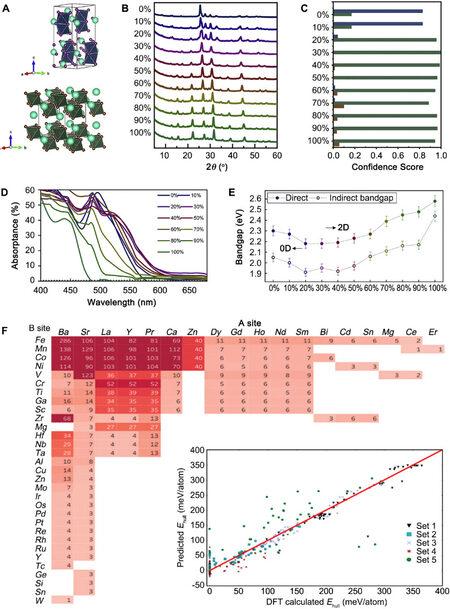
Figure 6. (A) Structure of Cs3(Bi1-xSbx)2(I1-xBrx)9. (B) XRD data of Cs3(Bi1-xSbx)2(I1-xBrx)9 with different x. (C) Confidence score on the dimensionality of perovskite. (D) Absorbance data of Cs3(Bi1-xSbx)2(I1-xBrx)9 with different x. (E) Bandgap “bowing” phenomenon is caused by the doping coefficient. Copyright from Elsevier[54]. (F) Heat map of 1929 perovskite structures and the prediction result of five test subsets. Copyright from Elsevier[55]. DFT: Density functional theory.
Other properties correlated to product performance can also be learned and predicted, such as the stability of perovskite oxides. Stability is vital as it affects the PM operating conditions in energy-related products. ML models can help improve PM synthesis by finding desirable structures with high stability. Li et al.[55] constructed an ML model to achieve this using a popular ML tool known as scikit-learn, an open-source package in Python under the BSD license. Their training data was a subset of the data from Jacobs et al.[56]. The key contribution of their work was that they found the correlation between stability and dataset features. They first found out that a higher cross-validation F1 score can be achieved if more features are used in training, which means a huge structural characteristic space behind the property. They then successfully predicted the stability of perovskites in five subsets, including 242 perovskite structures in different forms, and found the key features that affect the stability. Recursive feature elimination (RFE) was used to select the most relevant features by removing some insignificant features in the prediction each time. They also printed a heat map of all 1929 perovskites and their constituents, as shown in Figure 6F. The heat map shows that most perovskites are constituted with Ba, Sr, La, Y, Pr, and Ca at the A site and Fe, Mn, Co, and Ni at the B site. Li and colleagues also successfully predicted the stability of new perovskite compounds, like BaFe0.25V0.75O3 and La0.5Y0.5Co0.5Mn0.5O3, in high-activity solid oxide fuel cell (SOFC) cathodes.
In addition to the work of Li et al.[55], Schmidt et al.[57] also performed a stability prediction based on various ML methods and DFT calculations. Their dataset was collected from ~250,000 cubic perovskite systems, including all theoretically existing perovskites and antiperovskites. They found that the periodic table information (group, period, and number of valence electrons) is sufficient to predict the energy distance and convex hull with 140 meV/atom in MAE, which means that stability is highly related to the size of the crystal cell and valence electrons. Deng et al.[58] also predicted the stability of perovskites using specific descriptors in linear regression.
Similar approaches are also performed in predicting the conductivity of perovskites. Energy applications, especially batteries and fuel cells, rely on the conductivity of materials. Previously, conductivity was challenging to estimate without experiments. ML is now available for this task. Priya et al.[59] offered an ML regression and classification workflow to predict the conductivity of perovskites. The feature importance scores provided by their XGBoost model suggest that the electronegativity and atom mass of B site atoms, including dopants, are the most significant features. The atom mass and electronegativity are also periodicity-correlated features, matching the result of Schmidt et al.[57]. Important features help us to discover the influential factors of the activation energy (eV) and total conductivity (S cm-1) of stable perovskites of different charge carrier types. Figures 7A-F show the conductivity prediction results for proton (H), oxide (O), protonic electronic (H+e), oxide electronic (O+e), oxide protonic (O+H), and electronic (e) perovskite conductors. The desired perovskite will appear and show excellent application performance by combining different property predictions.
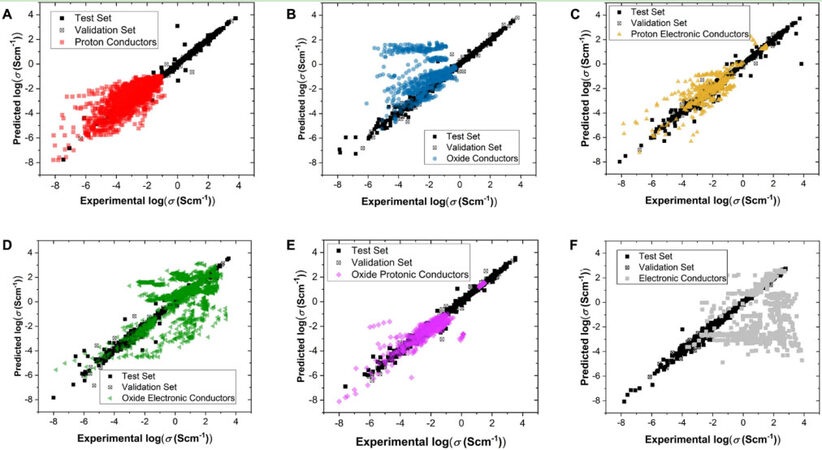
Figure 7. Predictions of conductivity compared to experimental values for (A) proton (H), (B) oxide (O), (C) protonic electronic (H+e), (D) oxide electronic (O+e), (E) oxide protonic (O+H), and (F) electronic (e) perovskite conductors. Copyright from Springer Nature[59].
In addition to PSCs, ML can aid other perovskite applications. Shen et al.[60] combined high-throughput calculations and ML to find electrostatic energy storage dielectrics. They designed an integrated phase-field model to understand the nanofiller effect on polymer nanocomposites. The output included effective permittivity, breakdown strength, and effective electrical conductivity. A total of 6615 calculated results were used as a dataset to train a BPNN model to estimate the energy storage capability. With this ML model, the authors found that parallel perovskite nanosheets can enhance the breakdown strength of polymer nanocomposites and they successfully fabricated a high-voltage endurance
Evaluating ML predictions through real products
The evaluation of PM performance is another important topic for energy applications. One cannot tell whether AI will promote PM performance before the product has been synthesized. Li et al.[63] built a two-model strategy based on LR, KNN, SVR, RF, and ANN models. The training data were extracted from 333 previous publications. The first model was used to predict the bandgaps of ABX3-type PMs. In contrast, the second model aimed to predict the open-circuit voltage (Voc), short-circuit current density (Jsc), fill factor (FF), and power conversion efficiency (PCE) of PSC devices. Eg, ΔH, and ΔL were used as inputs in the second model. The options of A, B, and X and the principle of ΔH and ΔL are shown in the right part of Figure 8A. The ANN showed the highest accuracy among all the ML results, giving 0.06 eV in RMSE and 0.97 R2 in bandgap prediction. For the PCE prediction with a true bandgap, 3.23% in RMSE and 0.80 in R2 were obtained. After training, they predicted and synthesized new films to evaluate the ML results.
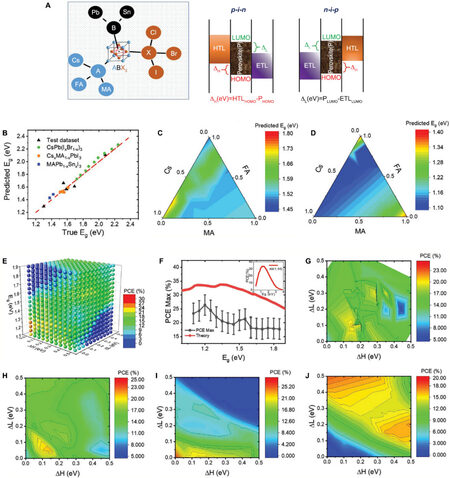
Figure 8. Model prediction and evaluation on synthesized perovskite thin film. (A) Description of A, B, and X in desired ABX3-type perovskite and explanation to ΔH and ΔL. (B) Predicted bandgap value versus experimental benchmark. (C, D) Bandgap rationalizes Cs/MA/FAPbI3 and Cs/MA/FASnI3 based on different Cs/MA/FA ratio. (E) Predicted PCE based on optimal values, implying that the highest PCE values are between 1.2 and 1.3 eV of bandgap with small ΔH and ΔL. (F) Comparison between PCE from Shockley and Queisser theory and maximum PCE from the model. (G, H) experimental ΔH, ΔL preference, and prediction map preference. (I, J) PCE map with ΔH and ΔL when Eg = 1.2 eV and Eg = 1.8 eV. Copyright from Wiley[63].
Doped perovskites, like CsxMA1-xPbI3, CsPb(IxBr1-x), and MAPb1-xSnxI3, were predicted and synthesized with measured Eg between 1.3 and 2.3 eV. Figure 8B shows the predicted Eg versus the experimentally tested Eg of these new PMs. Figures 8C and D show the bandgaps of perovskites with different MA, FA, and Cs ratios in Cs/MA/FAPbI3 and Cs/MA/FASnI3, which act as the interpretation of correlations between the A and B components and bandgap. The first model showed high consistency between the prediction and experimental benchmark and is thus capable of providing new PMs for PSCs. Under the instruction of the first model, PSCs were designed with certain Eg, ΔH, and ΔL.
Figure 8E shows the predicted PCEs based on these three values, implying that the highest PCE values are between 1.2 and 1.3 eV of Eg with small ΔH and ΔL. This result agrees with the Shockley and Queisser theory that the best PCE can be reached with materials having a Eg in the range of 1.15-1.35 eV. However, there are still differences between the theoretical and actual values. In Figure 8F, the red line denotes the theoretical limit and the grey line shows the maximum PCE. Figures 8G and H show the experimental ΔH and ΔL preference and predicted ΔH and ΔL with 1.5 eV Eg and PCE. The predicted value shows a high similarity with the choice of the authors. Figures 8I and J show that the highest PCE appears when
Gok et al.[64] developed a 2-step ML approach to predict the Eg and PCE using eight different perovskites compositions (RbCsFAMAPI, CsFAMAPI, CsFAPI, FAPI, MAPI, MAPI-Cl, FAPI + MAPBr, and FAMAPI-Br). This approach contains two RF models. The first model uses RF to predict the optical Eg. A total of 1437 UV-vis absorption plots were used as the training set for an RF model and the simulated results showed high accuracy, with all of the eight perovskites having an exceptional R2 > 0.99. Furthermore, the Tauc plots were used to estimate the Eg of experimental and predicted data. As a result, the predicted Eg values display a low deviation (< 1.4%) from the experimental results. After that, the second model was used to predict the current density-voltage (J-V) curves of PSCs, which can be used to calculate the PCE. The average R2 of the second model decreases to 0.9010 with a standard deviation of 0.0534. To verify the results, eight different perovskites were fabricated as absorber layers under the same laboratory conditions. Among them, MAPI-Cl-based PSCs were fabricated in a p-i-n configuration, while the rest were in n-i-p. This factor is not considered to simplify the model and the effects of the charge transporting layers and the interfaces on the device performance. Thus, the deviation between the measured PCE and predicted value of MAPI-Cl-based PSCs reached 3.176%, significantly larger than the others.
As Figure 9A shows, the experimental and ML results suggested that FAPI perovskite has the lowest
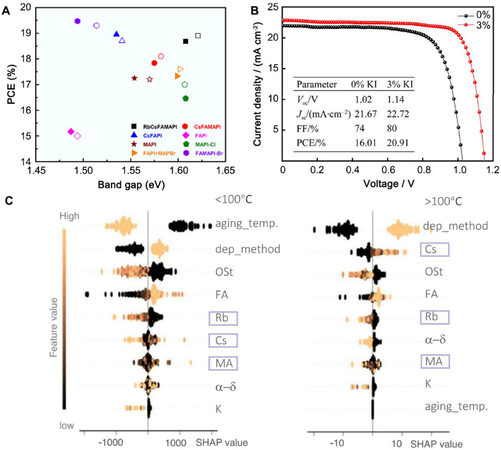
Figure 9. (A) PCE versus bandgap energy of eight perovskites. The filled symbols represent predicted results from ML, while the empty symbol represents experimental data. Copyright from Wiley[64]. (B) J-V curves of undoped and 3% KI-doped PSCs. Copyright from Springer Nature[65]. (C) Contribution of features under different temperatures. The feature importance is calculated from GBT regression and SHAP assessment, where aging_temp denotes aging temperature, dep_method denotes deposition method, Ost denotes over-stoichiometric with excess iodide, and α-δ denotes the probability of phase transition in humid air. The purple and orange color indicates low and high values of a given feature. Copyright from Springer Nature[66]. PCE: Power conversion efficiency; PSCs: perovskite solar cells; GBT: gradient boosting tree; SHAP: shapley additional explaining.
Another ML method to optimize KI doping in MAPbI3 solar cells was proposed by Jiang et al.[65]. They built a Gaussian process regression (GPR) model to predict the current density-voltage curve of KI-doped MAPbI3. The outcome suggested that 5% KI doping leads to the highest PCE. Three samples with doping concentrations of 3%, 5%, and 6% were synthesized to verify this result. The experimental result showed that the 3%-doped sample has a higher PCE than the 5%-doped one, which conflicts with the ML prediction. Thus, the new data were fed back to the training set for a second round of training. The prediction of this round showed that 3% is the optimal concentration, in agreement with previous experimental results. Seven different samples with doping concentrations of 0%, 1%, 2%, 3%, 5%, 8% and 10% were fabricated for further testing. It was proved that the 3% concentration KI doping provided the highest PCE, which illustrates that the ML model is reliable. In addition, the optimal PSC synthesized in this work achieves a higher FF, Voc and Jsc compared to the undoped MAPbI3 device and the PCE is improved from 16.01% to 20.91%, as shown in Figure 9B. This work demonstrated that ML is a reliable and powerful tool to optimize the doping method for hybrid perovskites.
Zhao et al.[66] developed an automated robotic system to search for stable perovskite solar cells. In this work, there was a learning cycle for the compositional screening of mixed-cation ABX3-type perovskites, where A denotes a monovalent cation (Cs, Rb, K, MA, or FA), B denotes lead, and X denotes a halide. Sixty-four compositions were selected and synthesized under different conditions. In total, over 1400 samples were synthesized and characterized by the robot. After that, a gradient boosting tree (GBT) model was used to explore the importance of each feature for stability at different temperatures. The results are shown in Figure 9C, which illustrates that the contributions of features are distinct under different temperatures, thus finding the optimal compositions for the different operating temperatures of PSCs. They further performed first-principles calculations for the perovskites to examine the thermal stability. Considering the T80 [the time for a 20% decay of photoluminescence (PL)] and thermal stability, MAxCs0.15-xFA0.85PbI3 PSCs were fabricated with an n-i-p structure. The average PCE of MAxCs0.15-xFA0.85PbI3 with x = 5% and 10% increased from 17.5% to 19.1% and 18.3% compared with the MA-free perovskite. The PCE loss of 5%-10% of MA devices is less than 5% under 85 °C after 1400 h, while the MA-free one suffers ~25%. The MAxCs0.15-xFA0.85PbI3-based device could even maintain 90% of its peak PCE value after 1800 h of continuous operation.
ACCELERATED SYNTHESIS FOR PEROVSKITES
Accelerated synthesis process through ML
Due to the complex parameter space of its structure, perovskite synthesis is a sophisticated process with high time costs and strict requirements for reaction conditions, especially for perovskite nanostructures[67]. This problem hinders the exploration of new perovskites because traditional trial-and-error requires vast amounts of experiments. Even though simulation-based methods like DFT could help estimate parameters, the expensive computation resources and long calculation time drastically reduce their practicality. After AI extends its application in the experimental field, it offers a highly efficient method to develop, characterize and optimize devices, saving time and effort by avoiding numerous manual experiments[31]. One of the popular methods in previous research was to use ML to guide or control the synthesis process.
For example, Braham et al.[68] used SVM classification and regression models to control the synthesis of perovskite halide nanoplatelets by determining the high-yielded quantum-confined CsPbBr3 nanoplatelets from the design space. Yang et al.[69] combined ML models with DFT calculations to obtain excellent double PMs from 16400 candidates, ideal for high-performing PSCs. They first used the gradient boosting decision tree (GBDT) model to predict the bandgap of 16400 candidates and then select proper structures for DFT calculations based on the bandgap, tolerance factor, octahedral factor, and atom at the X site. Finally, 61 possible structures were chosen, and the DFT results showed that ten of them fulfilled the requirement. To improve the stability of energy harvesting and conversion using halide perovskites, Sun et al.[70] developed a closed-loop Bayesian optimization framework to search stable composition of CsxMAyFA1-x-yPbI3. Only sampling 1.8% of the discretized compositional space, the model found an FA-rich and Cs-poor region centered with > 17-fold stability. The authors built an ML-based method using the ideal of high throughput experimentation (HTE) to synthesize and identify the lead-free perovskite composition with a Eg between 1.2 and 2.4 eV, which is desired for energy-harvesting applications[54]. This work finally investigated 75 different perovskite compositions spanning ABX3, A3B2X9, ABX4 and A2BIBIIIX6.
In recent years, with the rise of the above-mentioned AI-assisted synthesis methods, automated experimental systems inspired by AI have been proposed by researchers[71]. The automated experimental systems, integrating the concepts of HTE, robot automation systems, and ML models, significantly reduced the experimental time cost and improved the quality of reaction products. Typically, compared to only ML-based methods, these systems have a closed loop of experiment execution and self-learning to optimize the synthesis process[72]. Thus, they are more suitable for perovskite discovery. Kirman et al.[47] developed an ML-assisted perovskite discovery framework with automatic synthesis and automated characterization. The workflow is shown in Figure 10A. The framework includes two ML models: an image recognition model for crystal classification and a predictive regression model. A CNN was first trained with a dataset containing 25000 crystal images to distinguish between good crystal formation and no crystal formation. An ML regression model was then used to predict the likelihood of crystallization in the experimental space. The successful experiment rate doubled only after one experimental cycle with the classifier, distinctly avoiding the time-consuming synthesis process duplication. Additionally, they found a new structure (3-PLA)2PbCI4 that showed a solid blue emission using the framework.
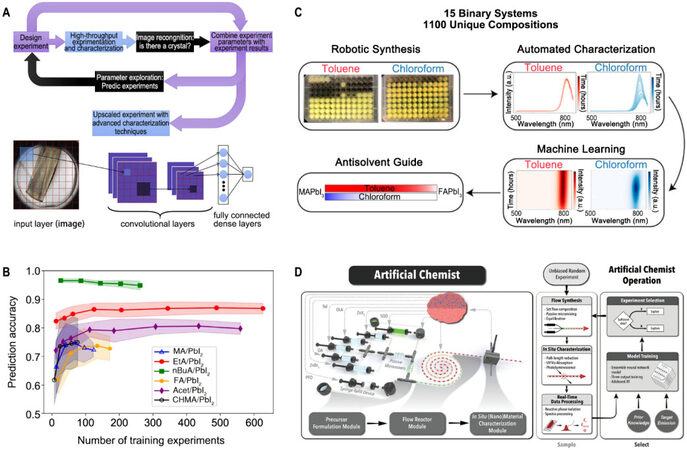
Figure 10. (A) Workflow for high-throughput synthesis of single-crystal perovskites and the image-recognition classification model. Copyright from Elsevier[47]. (B) Prediction accuracy vs. the number of training experiments for PUFK-SVM models of different crystallization systems. Solid lines show mean accuracy, and shaded bands indicate the standard deviation from five-fold CV results for each system. Copyright from ACS Publications[73]. (C) Workflow of ML-guide robot-based MHPs synthesis system. Copyright from ACS Publications[77]. (D) Schematic of the developed intelligent modular fluidic microprocessor for autonomous synthetic path discovery and optimization of colloidal QDs and the process flow diagram detailing its operation. Copyright from Wiley[78].
There are many examples of successfully-constructed automated platforms for the research and development of PMs. Li et al.[73] built a high-throughput robotic system for controlling the growth of metal halide perovskite crystals. They combined high-throughput experimentation and an ML model to build an automated perovskite synthesis platform, which could optimize the reaction parameters itself to obtain suitable crystals (> 0.1 mm) for single-crystal X-ray diffraction. The system records the experiment conditions and results. It can form a dataset to train specific binary classification models (SVM, k-NN, and RDF) to distinguish the high-quality single crystals. The accuracy of the model increased with the number of experiments, as shown in Figure 10B. Although this system has certain limitations in practice, it successfully carried out 8172 perovskite synthesis reactions ten times faster than human labor and discovered two novel perovskite species, AcetPbI3 and (CHMA)2PbI4.
A robotic system constructed by Chen et al.[74] automatically enabled the synthesis and characterization of perovskites, which helped them identify four perovskite compositions from 95 tested targets with an optical Eg ≈ 1.75 eV and sufficient stability. Another robotic-based system developed by Gu et al.[75] provided a deep insight into the antisolvent effect for lead halide perovskites. Higgins et al.[76] developed an automated perovskite discovery system to search for PMs with long-term stability. The system could synthesize perovskites and measure the PL spectra without an operator. With non-negative matrix factorization and Gaussian process regression, the system can determine the most stable region in the phase diagram by analyzing the photoluminescent behavior. They further utilized their system to investigate the effect of antisolvents on multicomponent metal halide perovskites (MHPs), which are used to fabricate high-quality MHP films[77]. Figure 10C shows how the robot synthesized 1100 compositions. The sample was doubled using two different antisolvents, namely, toluene and chloroform. The ML model then learned from the characterization data and interpreted that the selection of antisolvents would influence the photoluminescence behavior of MHPs. Epps et al.[78] designed an artificial chemist that could synthesize perovskite quantum dots (QDs) and learn and discover synthesis routes by itself. A pre-trained NNE model was used to form a closed loop, as shown in Figure 10D. The system could synthesize colloidal QDs and measure their PL quantum yield (PLQY), emission linewidth (EFWHM), and peak emission energy (EP), whilst recording the information on reaction flow and properties of QDs as training data for the synthesis route optimization in the NNE model to obtain the product with desirable properties. After 25 loops, the system obtained high-quality perovskite QDs within 1 meV of 11 target EP.
Indeed, ML-led automated laboratories offer a better perovskite synthesis solution to labor-intense trial-and-error exploration in the complex space of perovskite structures. Moreover, this ideal has been used for related studies like hole transport materials (HTMs) used for PSCs[79] and organic photovoltaics[80], thus boosting the energy harvesting ability of PMs. However, it is noteworthy that the startup cost of an ML-led automated laboratory is high.
Accelerated PM synthesis through cloud laboratories
Generally, each ML-based or automated experiment requires expensive hardware and computational resources, resulting in a limitation for studies. Cloud laboratories have thus become an ideal solution for digital chemical experiments. Since the concept of cloud computing was established about 20 years ago, it has become a buzzword in the IT industry[81]. It is an on-demand self-service model for broad network access to a pool of computing resources, including storage, memory, and processing, which can be rapidly provisioned and released[82]. Nowadays, well-built cloud-based laboratories exist, such as Transcriptic and Emerald Cloud Laboratories[83]. Inspired by these achievements, material scientists have developed cloud labs for perovskite discovery.
In 2020, Li et al.[84] constructed an intelligent cloud lab for optically active perovskite nanocrystal (IPNC) discovery, which is an update of their previous work[85]. Figures 11A and B illustrate the architecture of this cloud lab. A central platform, materials acceleration operating system in the cloud (MAOSIC), is used to connect the automated experimental system to cloud servers. The MAOSIC platform works as a multi-functional interface that allows users to control the hardware, obtain experiment data, observe experiment status and help the system optimize the reaction parameters. The wireless 5G network and an encrypted tunnel were applied for data transmission regarding the stability and security problems[86]. The users can only access the server by the key-built socket shell (SSH) tunnel, thereby improving security and efficiency. For the experiment part, SNOBFIT algorithms combine random search and gradient descent method is used to explore the high circular dichroism (CD) intensity region in the synthesis parameter space (temperature and concentration). Compared to the automated system introduced in the above section, cloud laboratories overcome the limitations of equipment and resources, offering users a more straightforward method to experiment while keeping the advantages of high operation speed, self-learning, and high accuracy. It has laid a solid foundation for the application of AI-assisted perovskite research systems. Novel PMs sometimes show interesting phenomena and extend the PM application field. It was the first time that chirality absorbance was found in an inorganic PM, and the PM was discovered and synthesized by an automatic MAOSIC system. This work shows that with the help of AI and robotic systems, more novel energy materials, especially PMs, are waiting for discovery and will contribute to human lives in the future. All abbreviations used in this review are collected in Table 2 for reference.
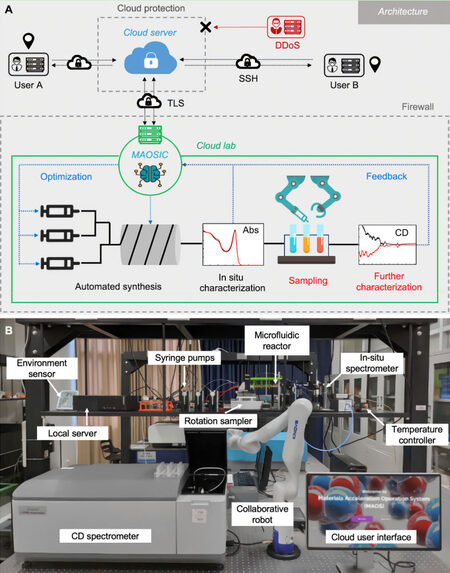
Figure 11. (A) Cloud lab architecture: the central platform MAOSIC allowed remoted users to control the (B) automated robot system through the cloud server. Copyright from Springer Nature[84]. SSH: Socket shell; CD: circular dichroism.
Abbreviation used in this review
| Abbreviation | Full meaning |
| AI | Artificial Intelligence |
| ANN | Artificial Neural Network |
| AUC | Area under the ROC curve |
| CD | Circular Dichroism |
| CGCNN | Crystal Graph Convolutional Neural Network |
| CMR | Computational Materials Repository |
| CNN | Convolutional Network |
| COD | Crystallography Open Database |
| CSD | Cambridge Structural Database |
| DBSCAN | Density-based spatial clustering of applications with noise |
| DFT | Density Functional Theory |
| EFWHM | Emission Linewidth |
| Eg | Bandgap Energy |
| EP | Peak Emission Energy |
| ETL | Electron Transport Layer |
| FF | Fill Factor |
| GA | Genetic Algorithm |
| GBDT | Gradient Boosting Decision Tree |
| GBRT | Gradient Boosting Regression Tree |
| GBT | Gradient Boosting Tree |
| GPR | Gaussian Process Regression |
| HOIP | Hybrid Organic-Inorganic Perovskite |
| HTE | High Throughput Experimentation |
| HTL | Hole Transport Layer |
| HTM | Hole Transport Material |
| ICSD | Inorganic Crystal Structure Database |
| IPNC | Intelligent Cloud Lab for Optically Perovsktie Nanocrystals |
| Jsc | Short-circuit Current |
| k-fold CV | l-fold Cross-Validation |
| LOOCV | Leave-One-Out Cross-Validation |
| MAE | Mean Absolute Error |
| MAOSIC | Materials Acceleration Operation System in Cloud |
| MHP | Metal Halide Perovskite |
| ML | Machine Learning |
| MSE | Mean Square Error |
| NN | Neural Network |
| OQMD | Open Quantum Materials Database |
| PCA | Principal Component Analysis |
| PCE | Power Conversion Efficiency |
| PL | Photoluminescence |
| PLQY | PL Quantum Yield |
| PM | Perovskite Materialss |
| PSC | Perovskite Solar Cell |
| QD | Quantum Dot |
| R | Regression correlation coefficient |
| R2 | coefficient of determination |
| RF | Random Forest |
| RFE | Recyrsuve Feature Elimination |
| RMSE | Root Mean Square Error |
| ROC | Receiver Operating Characteristic Curve |
| SHAP | Shapley Additional Explaining |
| SOFC | Solid Oxide Fuel Cell |
| SSH | Socket Shell |
| SVD | Singular Value Decomposition |
| SVM | Support Vector Machine |
| SVR | Support Vector Regressor |
| Tc | Curie temperature |
| Voc | Open-circuit Voltage |
| VASP | Vienna ab initio Simulation Package |
CONCLUSION AND OUTLOOK
This review has summarized the perspectives of AI-assisted discovery methods of PMs and reviewed how AI improves PMs in energy harvesting devices. The effects of AI can be mainly divided into three parts: property prediction, synthesis acceleration, and device design. We list AI assistances in different PM types, including ABX3, A3B2X9, ABX4, A2BIBIIIX6, AXB1-XCX3 and ABXC1-XX3. In PM research and development, AI shares the tasks of theoreticians, experimental platforms, and practical operators, which ML, cloud laboratories, and robotic systems respectively realize. The usage of ML can be divided into four parts: singe-model ML method, multi-model cooperation, NNs, and physics computation-assisted ML. The two approaches, DFT and GW, help organize the training for the last type. The critical points for a successful ML model are new training data, feature engineering, and model selection. ML has already discovered new PMs with desired properties, which show outstanding performance in devices, and more preciously, the interpretable ML models show theoretical consistency. Cloud laboratories remove the barriers of the limited research budget, while robotic systems commit to the precise synthesis of specific PMs. Due to the complexity and diversity of PMs and device architecture, the trend of AI-assisted PM discovery and improvement will be unstoppable in the future.
Despite these achievements, there still exist some problems in AI-assisted PM applications. Along with reliable solutions to the following challenges, PM discovery and applications should become more integral for energy-harvesting missions:
1. Currently, ML and NN procedures in PMs and correlated devices lack data. Many present ML models for PMs use only thousands of perovskite structures with properties. Thousands are small compared to ML for general inorganic and organic structures, like oxides and specific molecules. Data shortage may come from the limited options of A, B, and X in the perovskite formula, although doping can enrich the diversity. DFT calculations are also costly because the unit cell of perovskite usually contains dozens of atoms, and detailed parameters need to be set for high accuracy. For inorganic-organic perovskites, it is not easy to calculate specific properties using DFT or GW. To improve prediction performance, enlarging the perovskite database is essential.
2. Detailed interpretation and consistency with theory are essential. This problem is less severe in ML methods, but NNs are black boxes. Although NNs can almost restore the relationship between PM features and properties (large R2 and small MAE and RMSE), its interpretation is not implementable. Despite some visualization methods to see the feature weights of each layer, the contribution to the final prediction value is still hard to interpret. Physical-endorsed ML[87] and NNs[88] partially solve this problem, contributing to perovskite AI approaches.
3. The improvements in accuracy should occur along with the synthesis of new structures and characterization methods. ML approaches are commonly used for property predictions, like bandgaps, thermodynamic stability, and absorbance. New structures should be predicted and synthesized to accelerate new PM discovery, besides improving the scores on prediction tasks. Meanwhile, characterization methods should be updated to evaluate new PM performance. It is also encouraged to construct devices based on new PMs and test the improvements.
DECLARATIONS
Authors’ contributionsConceptualization: Zhu X, Zhao Y
Data collection: Liang J, Hu L
Data analysis & visualization: Liang J, Wu T
Manuscript preparation: Liang J, Wu T, Wang Z, Yu Y
Review & editing: Liang J, Zhu X, Zhao Y
Project administration & Funding acquisition: Zhu X, Zhao Y
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work is supported by the National Natural Science Foundation of China (grant nos. 22075240, 22179031, 21805234), the Shenzhen Fundamental Research Foundation (JCYJ20210324142213036, JCYJ20180508162801893) and Funding from the Shenzhen Institute of Artificial Intelligence and Robotics for Society (AIRS) is appreciated.
Conflicts of interestAll authors declare that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Perera ATD, Nik VM, Chen D, Scartezzini J, Hong T. Quantifying the impacts of climate change and extreme climate events on energy systems. Nat Energy 2020;5:150-9.
2. Capellán-pérez I, Mediavilla M, de Castro C, Carpintero Ó, Miguel LJ. Fossil fuel depletion and socio-economic scenarios: an integrated approach. Energy 2014;77:641-66.
3. Bashir T, Ismail SA, Song Y, et al. A review of the energy storage aspects of chemical elements for lithium-ion based batteries. Energy Mater 2021;1:100019.
4. Xiao Y, Xu R, Xu L, Ding J, Huang J. Recent advances on anion-derived sei for fast-charging and stable lithium batteries. Energy Mater 2021;1:100013.
5. Alias N, Mohamad AA. Advances of aqueous rechargeable lithium-ion battery: A review. J Power Sources 2015;274:237-51.
6. Li C, Zhang X, Zhu Y, et al. Modulating the lithiophilicity at electrode/electrolyte interface for high-energy Li-metal batteries. Energy Mater 2021;1:100017.
7. Zhang L, Chen Y. Electrolyte solvation structure as a stabilization mechanism for electrodes. Energy Mater 2021;1:100004.
8. Ruan J, Sun H, Song Y, et al. Constructing 1D/2D interwoven carbonous matrix to enable high-efficiency sulfur immobilization in Li-S battery. Energy Mater 2021;1:100018.
9. Chen X, Zhao J, Li G, Zhang D, Li H. Recent advances in photocatalytic renewable energy production. Energy Mater 2022;2:200001.
10. Yahya N, Aziz F, Jamaludin N, et al. A review of integrated photocatalyst adsorbents for wastewater treatment. J Environ Chem Eng 2018;6:7411-25.
11. Wang X, Ou P, Ozden A, et al. Efficient electrosynthesis of n-propanol from carbon monoxide using a Ag-Ru-Cu catalyst. Nat Energy 2022;7:170-6.
12. Vangari M, Pryor T, Jiang L. Supercapacitors: review of materials and fabrication methods. J Energy Eng 2013;139:72-9.
13. He C, Cheng J, Liu Y, Zhang X, Wang B. Thin-walled hollow fibers for flexible high energy density fiber-shaped supercapacitors. Energy Mater 2021;1:100010.
14. Zhang L, Hu X, Wang Z, Sun F, Dorrell DG. A review of supercapacitor modeling, estimation, and applications: a control/management perspective. Renew Sust Energ Rev 2018;81:1868-78.
15. Yang M, Wei W, Zhou X, Wang Z, Duan C. Non-fused ring acceptors for organic solar cells. Energy Mater 2021;1:100008.
16. Zhang C, Liang S, Liu W, et al. Ti1-graphene single-atom material for improved energy level alignment in perovskite solar cells. Nat Energy 2021;6:1154-63.
17. Dodds PE, Staffell I, Hawkes AD, et al. Hydrogen and fuel cell technologies for heating: a review. Int J Hydrog Energ 2015;40:2065-83.
18. Lu Y, Zhu B, Shi J, Yun S. Advanced low-temperature solid oxide fuel cells based on a built-in electric field. Energy Mater 2021;1:100007.
19. Zhu B, Mi Y, Xia C, et al. Nano-scale view into solid oxide fuel cell and semiconductor membrane fuel cell: material and technology. Energy Mater 2021;1:100002.
20. Zhang X, Liu G, Zhou K, et al. Enhancing cycle life of nickel-rich LiNi0.9Co0.05Mn0.05O2 via a highly fluorinated electrolyte additive - pentafluoropyridine. Energy Mater 2021;1:100005.
21. Wang Y, Xu H, Zhong J, et al. Hierarchical Ni/Co-based oxynitride nanoarrays with superior lithiophilicity for high-performance lithium metal anode. Energy Mater 2021;1:100012.
22. Yang C, Gao N, Wang X, et al. Phosphate boosting stable efficient seawater splitting on porous NiFe (oxy)hydroxide@NiMoO4 Core-Shell micropillar electrode. Energy Mater 2021;1:100015.
23. Guan Z, Wu Y, Wang P, et al. Perovskite photocatalyst CsPbBr3-xIx with a bandgap funnel structure for H2 evolution under visible light. Appl Catal B 2019;245:522-7.
24. Cui P, Qu S, Zhang Q, et al. Perovskite homojunction solar cells: opportunities and challenges. Energy Mater 2021;1:100014.
25. Mei A, Li X, Liu L, et al. A hole-conductor-free, fully printable mesoscopic perovskite solar cell with high stability. Science 2014;345:295-8.
26. Kim HS, Jang IH, Ahn N, et al. Control of I-V hysteresis in CH3NH3PbI3 perovskite solar cell. J Phys Chem Lett 2015;6:4633-9.
27. Lu J, Li Y. Perovskite-type Li-ion solid electrolytes: a review. J Mater Sci: Mater Electron 2021;32:9736-54.
28. Choudhary K, Bercx M, Jiang J, Pachter R, Lamoen D, Tavazza F. Accelerated discovery of efficient solar-cell materials using quantum and machine-learning methods. Chem Mater 2019;31:5900-8.
30. Wang Q, Phung N, Di Girolamo D, Vivo P, Abate A. Enhancement in lifespan of halide perovskite solar cells. Energy Environ Sci 2019;12:865-86.
31. Srivastava M, Howard JM, Gong T, Rebello Sousa Dias M, Leite MS. Machine learning roadmap for perovskite photovoltaics. J Phys Chem Lett 2021;12:7866-77.
32. Pilania G, Mannodi-Kanakkithodi A, Uberuaga BP, Ramprasad R, Gubernatis JE, Lookman T. Machine learning bandgaps of double perovskites. Sci Rep 2016;6:19375.
33. Im J, Lee S, Ko T, Kim HW, Hyon Y, Chang H. Identifying Pb-free perovskites for solar cells by machine learning. npj Comput Mater 2019:5.
34. Tao Q, Xu P, Li M, Lu W. Machine learning for perovskite materials design and discovery. npj Comput Mater 2021:7.
35. Jain A, Ong SP, Hautier G, et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Materials 2013;1:011002.
36. Saal JE, Kirklin S, Aykol M, Meredig B, Wolverton C. Materials design and discovery with high-throughput density functional theory: the open quantum materials database (OQMD). JOM 2013;65:1501-9.
37. Castelli IE, Landis DD, Thygesen KS, et al. New cubic perovskites for one- and two-photon water splitting using the computational materials repository. Energy Environ Sci 2012;5:9034.
38. Hellenbrandt M. The inorganic crystal structure database (ICSD) - present and future. Crystallography Reviews 2014;10:17-22.
39. Curtarolo S, Setyawan W, Hart GL, et al. AFLOW: an automatic framework for high-throughput materials discovery. Comput Mater Sci 2012;58:218-26.
40. Groom CR, Bruno IJ, Lightfoot MP, Ward SC. The cambridge structural database. Acta Crystallogr B Struct Sci Cryst Eng Mater 2016;72:171-9.
41. Gražulis S, Daškevič A, Merkys A, et al. Crystallography Open Database (COD): an open-access collection of crystal structures and platform for world-wide collaboration. Nucleic Acids Res 2012;40:D420-7.
42. Jain A, Montoya J, Dwaraknath S, et al. The materials project: Accelerating materials design through theory-driven data and tools. In: Andreoni W, Yip S, editors. Handbook of Materials Modeling: Methods: Theory and Modeling. Springer; 2020. pp. 1751-84. .
43. Oviedo F, Ren Z, Sun S, et al. Fast and interpretable classification of small X-ray diffraction datasets using data augmentation and deep neural networks. npj Comput Mater 2019:5.
44. Xu Q, Li Z, Liu M, Yin WJ. Rationalizing perovskite data for machine learning and materials design. J Phys Chem Lett 2018;9:6948-54.
45. Zhai X, Chen M, Lu W. Accelerated search for perovskite materials with higher Curie temperature based on the machine learning methods. Comput Mater Sci 2018;151:41-8.
46. Xie T, Grossman JC. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys Rev Lett 2018;120:145301.
47. Kirman J, Johnston A, Kuntz DA, et al. Machine-learning-accelerated perovskite crystallization. Matter 2020;2:938-47.
48. Saidi WA, Shadid W, Castelli IE. Machine-learning structural and electronic properties of metal halide perovskites using a hierarchical convolutional neural network. npj Comput Mater 2020:6.
49. Vicente N, Garcia-Belmonte G. Methylammonium lead bromide perovskite battery anodes reversibly host high li-ion concentrations. J Phys Chem Lett 2017;8:1371-4.
50. Liu M, Johnston MB, Snaith HJ. Efficient planar heterojunction perovskite solar cells by vapour deposition. Nature 2013;501:395-8.
51. He Q, Gu H, Zhang D, Fang G, Tian H. Theoretical analysis of effects of doping MAPbI into p-n homojunction on several types of perovskite solar cells. Optical Materials 2021;121:111491.
52. Odabaşı Ç, Yıldırım R. Performance analysis of perovskite solar cells in 2013-2018 using machine-learning tools. Nano Energy 2019;56:770-91.
53. Ye M, Hong X, Zhang F, Liu X. Recent advancements in perovskite solar cells: flexibility, stability and large scale. J Mater Chem A 2016;4:6755-71.
54. Sun S, Hartono NT, Ren ZD, et al. Accelerated development of perovskite-inspired materials via high-throughput synthesis and machine-learning diagnosis. Joule 2019;3:1437-51.
55. Li W, Jacobs R, Morgan D. Predicting the thermodynamic stability of perovskite oxides using machine learning models. Computational Materials Science 2018;150:454-63.
56. Jacobs R, Mayeshiba T, Booske J, Morgan D. Material discovery and design principles for stable, high activity perovskite cathodes for solid oxide fuel cells. Adv Energy Mater 2018;8:1702708.
57. Schmidt J, Shi J, Borlido P, Chen L, Botti S, Marques MAL. Predicting the thermodynamic stability of solids combining density functional theory and machine learning. Chem Mater 2017;29:5090-103.
58. Deng Q, Lin B. Automated machine learning structure-composition-property relationships of perovskite materials for energy conversion and storage. EM 2021; doi: 10.20517/energymater.2021.10.
59. Priya P, Aluru NR. Accelerated design and discovery of perovskites with high conductivity for energy applications through machine learning. npj Comput Mater 2021:7.
60. Shen Z, Bao Z, Cheng X, et al. Designing polymer nanocomposites with high energy density using machine learning. npj Comput Mater 2021:7.
61. Kim C, Pilania G, Ramprasad R. Machine learning assisted predictions of intrinsic dielectric breakdown strength of ABX3 perovskites. J Phys Chem C 2016;120:14575-80.
62. Xu P, Chang D, Lu T, Li L, Li M, Lu W. Search for ABO3 type ferroelectric perovskites with targeted multi-properties by machine learning strategies. J Chem Inf Model 2021; doi: 10.1021/acs.jcim.1c00566.
63. Li J, Pradhan B, Gaur S, Thomas J. Predictions and strategies learned from machine learning to develop high-performing perovskite solar cells. Adv Energy Mater 2019;9:1901891.
64. Gok EC, Yildirim MO, Haris MPU, et al. Predicting perovskite bandgap and solar cell performance with machine learning. Solar RRL 2022;6:2100927.
65. Jiang S, Wu C, Li F, et al. Machine learning (ML)-assisted optimization doping of KI in MAPbI3 solar cells. Rare Met 2021;40:1698-707.
66. Zhao Y, Zhang J, Xu Z, et al. Discovery of temperature-induced stability reversal in perovskites using high-throughput robotic learning. Nat Commun 2021;12:2191.
67. Xu X, Wang X. Perovskite nano-heterojunctions: synthesis, structures, properties, challenges, and prospects. Small Structures 2020;1:2000009.
68. Braham EJ, Cho J, Forlano KM, Watson DF, Arròyave R, Banerjee S. Machine learning-directed navigation of synthetic design space: a statistical learning approach to controlling the synthesis of perovskite halide nanoplatelets in the quantum-confined regime. Chem Mater 2019;31:3281-92.
69. Yang Z, Liu Y, Zhang Y, et al. Machine learning accelerates the discovery of light-absorbing materials for double perovskite solar cells. J Phys Chem C 2021;125:22483-92.
70. Sun S, Tiihonen A, Oviedo F, et al. A data fusion approach to optimize compositional stability of halide perovskites. Matter 2021;4:1305-22.
71. Ahmadi M, Ziatdinov M, Zhou Y, Lass EA, Kalinin SV. Machine learning for high-throughput experimental exploration of metal halide perovskites. Joule 2021;5:2797-822.
72. Häse F, Roch LM, Aspuru-guzik A. Next-generation experimentation with self-driving laboratories. Trends in Chemistry 2019;1:282-91.
73. Li Z, Najeeb MA, Alves L, et al. Robot-accelerated perovskite investigation and discovery. Chem Mater 2020;32:5650-63.
74. Chen S, Hou Y, Chen H, et al. Exploring the stability of novel wide bandgap perovskites by a robot based high throughput approach. Adv Energy Mater 2018;8:1701543.
75. Gu E, Tang X, Langner S, et al. Robot-based high-throughput screening of antisolvents for lead halide perovskites. Joule 2020;4:1806-22.
76. Higgins K, Valleti SM, Ziatdinov M, Kalinin SV, Ahmadi M. Chemical robotics enabled exploration of stability in multicomponent lead halide perovskites via machine learning. ACS Energy Lett 2020;5:3426-36.
77. Higgins K, Ziatdinov M, Kalinin SV, Ahmadi M. High-throughput study of antisolvents on the stability of multicomponent metal halide perovskites through robotics-based synthesis and machine learning approaches. J Am Chem Soc 2021;143:19945-55.
78. Epps RW, Bowen MS, Volk AA, et al. Artificial chemist: an autonomous quantum dot synthesis bot. Adv Mater 2020;32:e2001626.
79. MacLeod BP, Parlane FGL, Morrissey TD, et al. Self-driving laboratory for accelerated discovery of thin-film materials. Sci Adv 2020;6:eaaz8867.
80. Langner S, Häse F, Perea JD, et al. Beyond ternary OPV: high-throughput experimentation and self-driving laboratories optimize multicomponent systems. Adv Mater 2020;32:e1907801.
81. Wang L, von Laszewski G, Younge A, et al. Cloud computing: a perspective study. New Gener Comput 2010;28:137-46.
82. Mell P and Grance T, The NIST definition of cloud computing, 2011. Available from: http://faculty.winthrop.edu/domanm/csci411/Handouts/NIST.pdf [Last accessed on 13 May 2022].
84. Li J, Li J, Liu R, et al. Autonomous discovery of optically active chiral inorganic perovskite nanocrystals through an intelligent cloud lab. Nat Commun 2020;11:2046.
85. Li J, Lu Y, Xu Y, et al. AIR-chem: authentic intelligent robotics for chemistry. J Phys Chem A 2018;122:9142-8.
86. Dillon T, Wu C and Chang E. Cloud computing: issues and challenges. 2010 24th IEEE International Conference on Advanced Information Networking and Applications 2010:27-33.
87. Liang J, Zhu X. Phillips-inspired machine learning for band gap and exciton binding energy prediction. J Phys Chem Lett 2019;10:5640-6.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Liang J, Wu T, Wang Z, Yu Y, Hu L, Li H, Zhang X, Zhu X, Zhao Y. Accelerating perovskite materials discovery and correlated energy applications through artificial intelligence. Energy Mater 2022;2:200016. http://dx.doi.org/10.20517/energymater.2022.14
AMA Style
Liang J, Wu T, Wang Z, Yu Y, Hu L, Li H, Zhang X, Zhu X, Zhao Y. Accelerating perovskite materials discovery and correlated energy applications through artificial intelligence. Energy Materials. 2022; 2(3): 200016. http://dx.doi.org/10.20517/energymater.2022.14
Chicago/Turabian Style
Liang, Jiechun, Tingting Wu, Ziwei Wang, Yunduo Yu, Linfeng Hu, Huamei Li, Xiaohong Zhang, Xi Zhu, Yu Zhao. 2022. "Accelerating perovskite materials discovery and correlated energy applications through artificial intelligence" Energy Materials. 2, no.3: 200016. http://dx.doi.org/10.20517/energymater.2022.14
ACS Style
Liang, J.; Wu T.; Wang Z.; Yu Y.; Hu L.; Li H.; Zhang X.; Zhu X.; Zhao Y. Accelerating perovskite materials discovery and correlated energy applications through artificial intelligence. Energy Mater. 2022, 2, 200016. http://dx.doi.org/10.20517/energymater.2022.14
About This Article
Copyright

Data & Comments
Data


 Cite This Article 44 clicks
Cite This Article 44 clicks

 Like This Article 21
likes
Like This Article 21
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.