
Designing the next generation of symmetrical organic redox flow batteries using helical carbocations
 0
0
Abstract
In recent years, non-aqueous fully organic Redox Flow Batteries (RFBs) have displayed potential in broadening the electrochemical window and enhancing energy density in RFBs by relying on redox-active organic molecules to provide improved sustainability in comparison to metal-based charge carriers. Of particular interest, systems that rely on a single bipolar redox molecule (BRM) for their operation, known as symmetrical organic RFBs, have gained momentum as the utilization of a BRM eliminates membrane crossover issues, thus extending the lifespan of electrical energy storage systems while reducing their cost. In this manuscript, we will present our contribution to this field through the design of tunable bipolar molecules within the helicene carbocation class. This particular type of BRM is synthetically very affordable and has proven to be highly modifiable and robust. Through the examination of 11 examples, we will demonstrate how an approach based on readily available electrochemical tools can be efficiently employed to generate and assess a library of compounds for future full flow RFB applications.
Keywords
INTRODUCTION
Meeting the ever-growing global energy demand is an urgent imperative, necessitating the development of energy-storage solutions. These solutions play a crucial role in facilitating the transition to renewable energy grid integration, decarbonizing our economy, and tackling the challenges posed by global warming[1-3]. In order to achieve the objectives outlined in the International Energy Agency roadmap[4], the emergence of a variety of metal-based battery solutions has been observed[5-8], notably marked by the widespread adoption of high-performance lithium-ion batteries[9-11]. However, as concerns about fire hazards linked to the accumulation of low-valent metal have risen and the current demand rapidly overtaking the availability of this metal[12,13], the industry is being directed toward safer, cost-effective, and scalable electricity energy storage (EES) solutions[14,15]. Redox flow batteries (RFBs) have emerged as a compelling choice for large-scale stationary applications within the EES domain, primarily because of their engineering adaptability and scalability enabled by the decoupling of power and capacity[16-19].
Currently, commercially established RFB technologies are based on the redox characteristics of transition metal ions in aqueous solutions (AqRFBs). Among these, the most extensively studied options are vanadium oxide and chromium and iron variants[20-24]. Nevertheless, several drawbacks hinder the widespread deployment of AqRFBs, such as the limited electrochemical stability window of water (1.5 V), low energy densities induced, high costs associated with ionic separators, environmental implications related to ore mining, management of substantial quantities of acid, and the inherent toxicity of certain compounds[25-27].To broaden the electrochemical window and enhance energy density under mild pH conditions, non-aqueous RFBs present a significant advancement[28-30]. They enable the use of Redox-active Organic Molecules (ROMs), demonstrating enhanced sustainability compared to transition metal-based redox-active materials[31]. ROMs have a lower global warming potential index, indicating an environmental impact one-fourth that of transition metal-based materials[32]. Moreover, they will ensure a consistent market price at one-fourth the cost per kilogram and can be sustainably sourced and tailored synthetically[31-33]. Most of reported Non-Aqueous fully-Organic RFBs (NAORFBs) exhibit an asymmetric configuration, using two distinct ROMs as catholyte and anolyte. This setup imposes selectivity requirements on the exchange membrane, leading to alternative avenues for chemical degradation and irreversible capacity fading[34,35]. Various effective approaches have been investigated to mitigate redox-active material crossover, including interface improved size selectivity[36,37], enhanced membrane composition[38-40], and eliminating the need for a membrane by adopting tandems of non-miscible redox electrolytes[41].
Concurrently, with the rise of innovation in the field, an elegant solution has emerged: drawing inspiration from vanadium RFBs (VRFBs) and their quasi-identical tank chemistry, symmetrical Organic RFBs (SORFBs) have gained prominence as the most promising approach for eliminating membrane crossover and prolonging EES lifespan[23,42-44]. The primary characteristic of SORFBs lies in their use of a singular bipolar redox molecule (BRM) on both sides of the cell[45]. This robust molecule features in its initial state -at least- three stable redox states, owing to entirely reversible reduction and oxidation processes. Consequently, these molecules can function as both anolyte and catholyte within a battery[46-48]. The resultant symmetrical RFB relies on identical solution components in each half-cell, offering notable advantages[42]. Firstly, using the same redox-active material reduces the chemical gradient of electroactive species, negating the necessity for highly selective membranes and effectively mitigating crossover. In the event of crossover, rather than enduring permanent contamination, SORFBs will undergo self-discharge[23]. Furthermore, when in the discharged state, there is no chemical or electrochemical gradient across the membrane, guaranteeing SORFBs to be stored indefinitely without contamination due to leakage or irreversible side reactions. Finally, recent research has demonstrated that capacity loss resulting from compound degradation can be reclaimed through regular polarity reversals, thus extending the battery lifespan[42,49-51]. Additionally, relying on a single charge carrier species could help achieve significant economies of scale while streamlining the supply chain for broad implementation. The recent surge in SORFB progress has inspired chemists to craft a varied array of BRMs, employing three molecular approaches to fashion novel redox-active compounds [Figure 1][45].
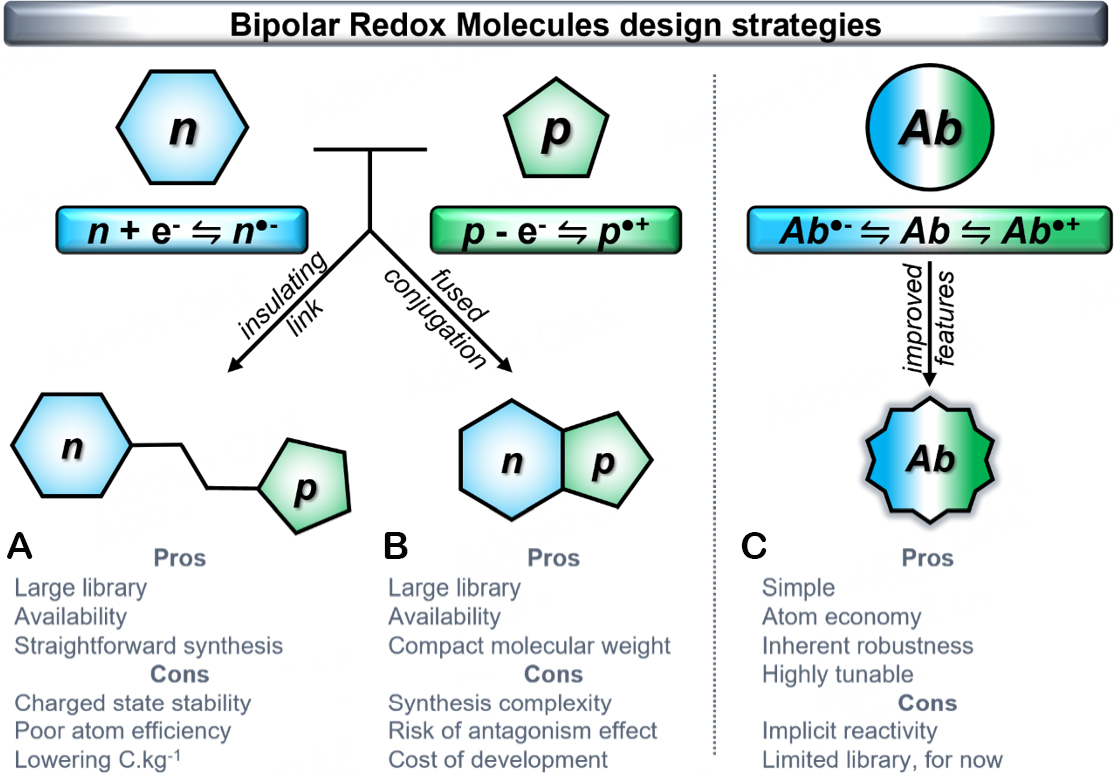
Figure 1. Three strategies to develop innovative bipolar redox molecules for the rise of Symmetrical Organic Redox Flow Battery. n corresponds to n-type molecules, p for p-type molecules, while Ab means for ambiphilic molecules. Examples of SORFBs based on strategy (A) could be found in the references[52-58], strategy (B) at[49,50,59,60] and strategy (C) at[51,61-64].
Redox-active organic materials are usually categorized as n-type or p-type based on their ability to either store or provide electrons in their neutral state during electrochemical reactions. n-type redox-active organic materials typically experience reduction from their neutral state, resulting in a negatively charged molecular state. In contrast, p-type organic materials undergo oxidation from their neutral state, yielding positively charged. Both ROMs must exhibit reversible redox events to be used. One straightforward method involves pairing an n-type moiety with a p-type moiety using a simple linker, most of the time insulating [Figure 1A]. The resulting molecule can function as a bipolar entity
An improved strategy for developing innovative BRMs enhances this design concept by electronically merging established n-type and p-type conjugated scaffolds within a single molecule [Figure 1B]. By closely integrating the conjugation of two aromatic materials, the electronic characteristics of each component are anticipated to synergistically influence the resulting electrochemical potentials. Rising the Egap while maintaining a reasonable molecular weight ensures a favorable capacity per unit weight. This route has proved successful, leading to the formation of closed-shell[49,58,60,67] and open-shell molecules[59,68] that exhibit improved properties compared to their parent units. Nonetheless, the synthetic access to such compounds can be a gradual and intricate process, involving multiple steps and introducing the risk of antagonism effects due to significant structural alteration of the initial n- and p- moieties. And, in contrast to method a, only a comprehensive electrochemical assessment of the final molecule can guarantee stability across all oxidation states (-●n|p
The latest approach shifts away from merging the electronic characteristics of n- or p-type molecules. Instead, it revolves around exploring existing literature for compounds that exhibit bipolar activity (Figure 1, -●Ab
Aligned with strategy (c) and contributing to the SORFB research field, our group chose to employ a specific category of BRMs within the carbenium ion class. Among the members of this carbocation family, certain compounds have demonstrated exceptional stability[72-74] and hold significant utility in coordination chemistry[75-77], small molecules activation[78,79], and, notably, organophotocatalysis[80-86]. And while a planar triangulenium motif has recently enabled the deployment of a robust SORFB model[87], it is the particularly rich and versatile chemistry of [4]helicenium ions that will be discussed in what follows[88]. Dimethoxyquinacridiniums (DMQA+) are sturdy helical carbenium ions, well-documented for their biological applications, photophysical features and electrochemical properties[89-96]. The reversible electrochemical oxidation and reduction processes involving these carbenium ions have been notably disclosed by Herse and Sørensen et al.[89,97]. In line with this, our group has recently published a study on the chemical synthesis, isolation, and characterization of neutral helicene radicals produced through chemical reduction of the corresponding carbocation precursors[98]. All of these preliminary efforts have enabled us to introduce a robust SORFB model based on a relatively straightforward N,N′-di-n-propyl-1,13-dimethoxyquinacridinium (nPrDMQA+) with the capability to operate in a poleless mode[99]. This research led to the publication of a second model featuring an enhanced DMQA+ framework through nitro group incorporation (nPrDMQANO2+), resulting in a significant boost in energy density at the expense of stability[100].
In this context, this work aims to demonstrate how the utilization of pre-established bipolar redox scaffolds facilitates effective screening. We propose a methodology for rapidly evaluating the properties and compatibility of new BRMs for SORFB deployment. This approach is illustrated through the examination of 11 differently substituted helicenium examples, leveraging our expertise on the DMQA+ core. The results will be showcased through electrochemical characterization using a three-electrode cell. A thorough investigation of electrochemical kinetic parameters will help qualify the effects induced by various substitutions and structure tethering. Lastly, the robustness of ROMs, which have demonstrated suitable parameters for use in the RFB field, will be evaluated through cycling in H-cells, also known as “static RFB”.
EXPERIMENTAL
General remarks
All solvents were purified by solvent purification system (SPS) or distillation over the drying agents indicated. Dried solvents and liquid reagents were transferred by oven-dried or hypodermic syringes. The supporting electrolyte salts, tetrabutylammonium hexafluorophosphate (TBAPF6) and tetrabutylammonium tetrafluoroborate (TBABF4), were recrystallized three times from ethanol and then dried at 80 °C for three days prior to use in the glovebox. All glassware or hardware has been dried in an oven at least 24 h prior to introduction in a glovebox.
Details of helicenium compound synthesis and nomenclature are available in Supplementary Table 1 and Supplementary Materials Synthesis part.
Electrochemistry
Electrochemical analyses were conducted inside an Argon-filled MBraun Unilab glovebox using a BioLogic SP-200 potentiostat/galvanostat and the EC-Lab® software (v11.50) from BioLogic Science Instruments. For convenience, potentials are expressed versus internal reference electrode AgNO3/Ag (Eref, 0.01 M AgNO3 in 0.1 M TBAPF6 in CH3CN).
Cyclic voltammetry and electrokinetic parameters determination
Cyclic voltammograms (CV) were obtained in a three-electrode electrochemical cell, which included a counter electrode (Ec) made of platinum wire, an AgNO3/Ag reference electrode (Eref), and a working electrode (Ew, 0.071 cm-2, CH Instrument, Inc.) composed of glassy carbon. Prior to each measurement, the working electrode was meticulously polished using aluminum oxide on polishing paper and anhydrous
Static RFB cycling
Bulk charge/discharge cycling was conducted within a homemade H-cell, where a porous glass frit served as the separator[101,102]. Reticulated vitreous carbon (RVC) electrodes were utilized as Ew and Ec, cut into standardized dimensions, and then disposed of at the conclusion of each experiment to eliminate the possibility of contamination. A Constant Current |5 mA| followed by a Constant Voltage Galvanostatic Charging with Potential Limitation (CCCV GCPL protocol) was applied via the RVC electrodes. Both chambers of the H-cell were filled with 5 mL of electrolyte/ROM and were continuously agitated with magnetic stir bars at 1,000 rpm. An equilibration period of two hours was observed prior to active charging and discharging. Details and dimensions are available in Supplementary Materials.
RESULTS AND DISCUSSION
The choice of the [4]helicenium motif as the core for structural modifications is related to its high durability and the numerous applications we have attributed to it within our research group. Indeed, the DMQA+ core, a stable carbocation in both air and water in its synthesized form, exhibits three stable redox states. The DMQA+ can thus be electrochemically reduced by one electron to form a neutral DMQA● radical at E1/2Red and conversely oxidized by one electron to form a DMQA●++ dication radical at E1/2Ox [Figure 2].
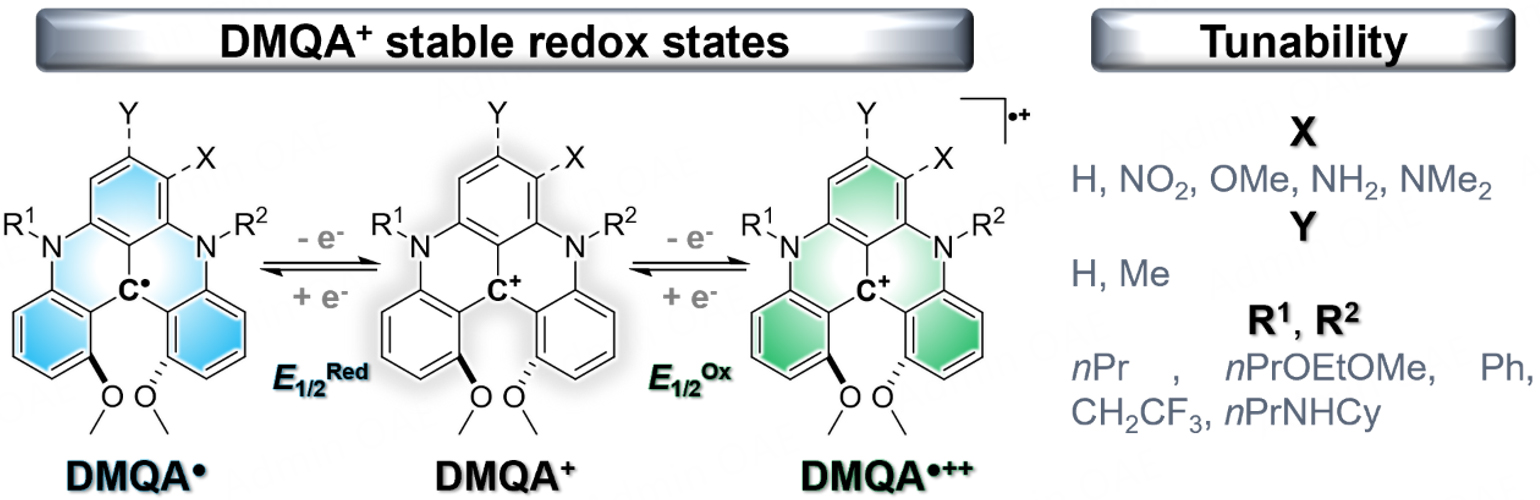
Figure 2. Stables redox states of DMQA+ core and structural modifications explored in this study.
The access and stability of these two states are crucial for obtaining a suitable BRM for symmetrical RFBs. Hence, all structural modifications explored must be compatible with the various oxidation states. Two variation types that showcase the extensive tunability of the DMQA+ motif can be investigated. Alterations of the aromatic core of the DMQA+ motif through positions X or Y with electrophilic or nucleophilic groups are feasible and detailed at various synthesis stages, whether initially on the tris-aryl carbenium synthon for Y or during the late functionalization stage for X [Supplementary Figure 1][94,100]. Changes in the nature of the R1 and R2 “arms” are almost invariably introduced during a double nucleophilic aromatic substitution [Supplementary Figure 1][103].
Considering the results obtained with the SORFB model based on nPrDMQA+[99], initially, we examined the consequences of modifying the X group through late-stage functionalization. The synthesis of these compounds had been well described and was achievable by adding only a few steps to the initial synthesis path[94]. The groups NO2, OMe, NH2, and NMe2, along with their influence on the electrochemical properties of the nPrDMQA+ scaffold as a reference, were thus studied in acetonitrile, our model solvent (Figure 3, nPrDMQA+ as black trace)[104,105]. The impact of the X-position substitution on the core has proved to be significant for the Egap and, thus, for attainable energy density[31]. As previously reported, the introduction of an electron-attracting nitro group contributes to shifting the potentials of the E1/2Red and
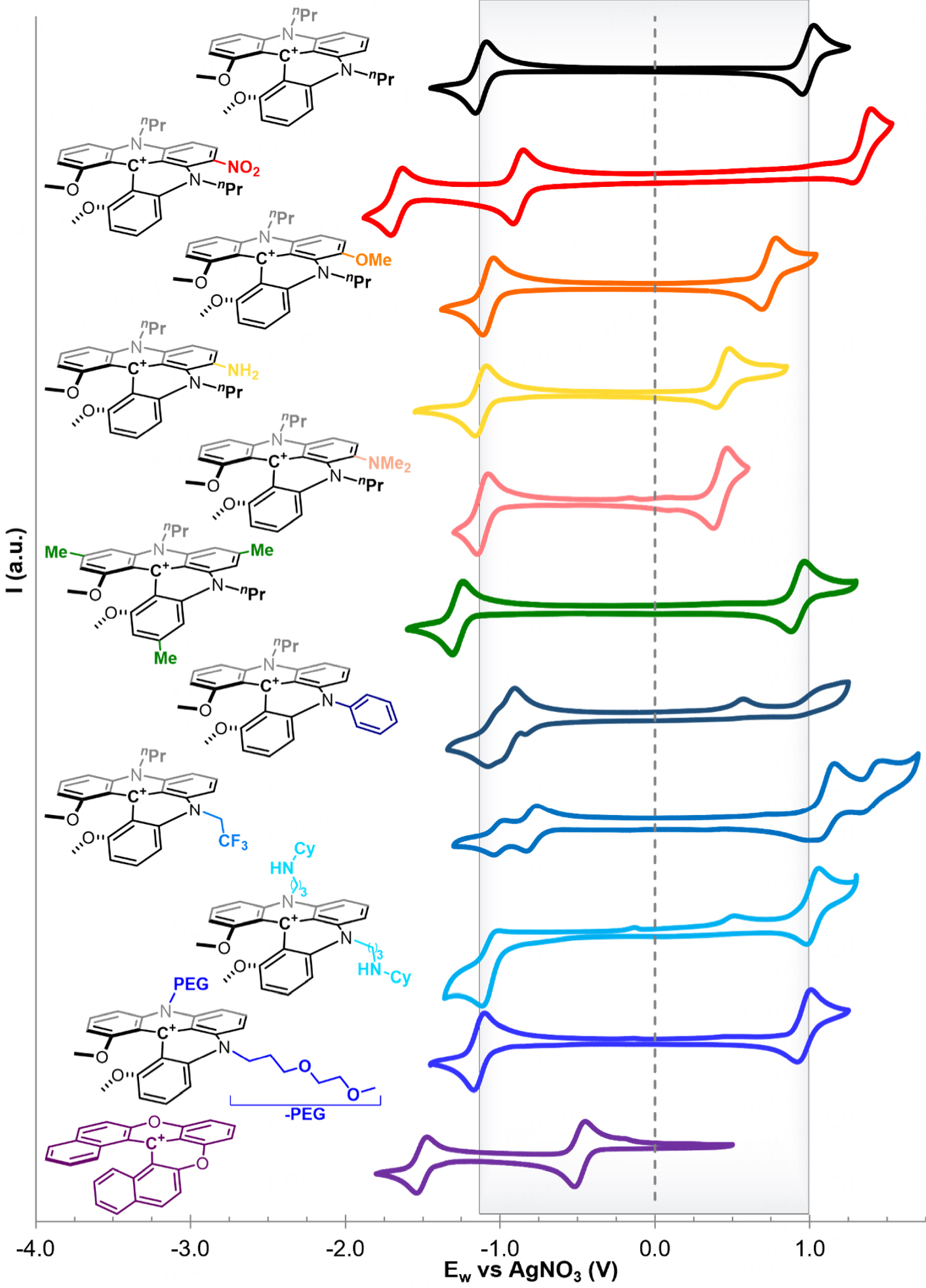
Figure 3. Cyclic voltammograms of 1 mM nPrDMQA+, nPrDMQANO2+,
When it comes to modifying the Y position of the aromatic core, options for late-stage functionalization are practically absent. The alteration of this position in the para of the carbocation center of tris-aryl carbenium synthons must take place during a preliminary step [Supplementary Figure 1]. Access to an electronically enriched scaffold through the introduction of OMe and NMe2 groups was unsuccessful[72,106]. Despite numerous attempts, the electron richness of acridinium moieties containing three σ- and π-donating groups, such as OMe and NMe2, in the para position inhibits the second aromatic nucleophilic substitution required to form the helicenium scaffold[83]. Fortunately, σ-donating groups such as methyl groups which were introduced in the para position to form the synthon tris(2,6-dimethoxy-4-methylphenyl)carbenium tetrafluoroborate did not hamper the synthesis of the helicenium analog DMQA(pMe3)+ (see
We then focus our interest on the nature of the arms carried by the DMQA+ scaffold and their impact on the electrochemical properties of the BRM. Through the introduction of a Ph group in R2 through a preliminary synthesis step, followed by adding an nPr group in R1, a dissymmetrical DMQA+ was obtained[103]. It was quickly observed that the presence of a Ph aromatic group, while not significantly altering the potential of the electrochemical events, severely disrupts their reversibility (Figure 3 deep blue trace). The E1/2Red was recorded at -0.99 V vs. AgNO3, a difference of +150 mV compared to the
Finally, the rich chemistry of heliceniums[109] prompted us to take an interest in dioxo[6]helicenium (labeled [6]helicene+ in Figure 3 and Table 1). [6]helicene+ is a parent helicenium of diaza[4]helicenium, the DMQAs presented thus far, devoid of R1/R2 arms where the bridging amino groups (NR) are replaced by a bridging oxygen atom. Expansion from [4] to [6]helicenes is provided by replacing the methoxy groups of the core with phenyl rings[110]. This [6]helicene+ presents two completely reversible reduction phenomena with a first reduction at E1/2Red = -0.48 V vs. AgNO3 at a much higher potential than the reduction of the DMQA+ model consistent with the less electron donating effect of the O vs. NR bridge groups (Figure 3 purple trace). However, no reversible electrochemical process is accessible in oxidation, consequently eliminating this molecule, as well as nPr/PhDMQA+, from use as a BRM, and these heliceniums will not be further discussed hereafter.
Summary of E1/2Red/Ox, Egap, diffusion parameters (D) and electron-transfer rate parameters (k0) of 1 mM R1/R2DMQAX/Y+ measured in 0.1 M TBAPF6 CH3CN. All potentials are expressed against AgNO3/Ag. Sets of 7 scan rates for electronic processes of each helicenium are available in Supplementary Figures 2-8.
| Compound | E1/2Red (V) | E1/2Ox (V) | EGap (V) | D (×10-6 cm2/s) | k0 (×10-2 cm/s) | ||
| E1/2Red | E1/2Ox | E1/2Red | E1/2Ox | ||||
| nPrDMQA+ | -1.14 | 0.98 | 2.12 | 9.49 | 9.99 | 2.65 | 2.04 |
| nPrDMQANO2+ | -0.89 | 1.35 | 2.24 | 5.57 | 9.87 | 3.69 | 0.79 |
| nPrDMQAOMe+ | -1.08 | 0.76 | 1.84 | 5.27 | 5.03 | 2.50 | 1.01 |
| nPrDMQANH2+ | -1.13 | 0.47 | 1.60 | 10.1 | 9.62 | 2.48 | 0.96 |
| nPrDMQANMe2+ | -1.12 | 0.42 | 1.54 | 5.70 | 4.05 | 2.23 | 0.66 |
| nPrDMQA (pMe)3+ | -1.27 | 0.92 | 2.19 | 5.82 | 4.12 | 3.43 | 0.293 |
| nPr/PhDMQA+ | -0.99 | irr | - | 4.01 | irr | 0.15 | irr |
| nPr/CH2CF3DMQA+ | -0.80 | 1.09* | 1.89* | 0.78 | 3.86 | 1.64 | 0.12 |
| (CyNHnPr)DMQA+ | -0.98‡ | 1.03‡ | 2.01 | 5.87 | 4.03 | 0.50 | 1.11 |
| PEGDMQA+† | -1.13 | 0.94 | 2.07 | 6.24 | 6.24 | 24.5 | 1.13 |
| [6]helicene+ | -0.48 | irr | - | 8.63 | irr | 2.45 | irr |
While the development of new BRMs for the deployment of SORFB is experiencing significant growth, it should be noted that a certain lack of coherence in the reporting and evaluation methods of their physicochemical specificities makes the comparison of new ROMs difficult[111]. However, certain kinetic parameters need to be highlighted. Thus, parameters such as diffusion (D) and electron-transfer rate (k0) will greatly determine the expected performances for using a ROM in SORFB. Through effective mass transport of the different oxidation states of the BRM from the bulk solution to the electrode surface, higher current density can be achieved while limiting overpotential. The importance of the parameter k0 lies in its description of the efficiency of electron transfer from the electrodes to the ROM. Its value range also indicates the reversibility of the electronic process, with complete reversibility above 10-1 cm·s-1, quasi-reversibility between 10-1 and 10-5 cm·s-1, and irreversibility below this range[112]. While having the highest possible values of D and k0 is indeed preferable, it should be noted that in the context of BRMs and due to the symmetrical aspect of SORFBs and the use of a single molecule, it is also important that the values corresponding to the reduction and oxidation processes are in the same order of magnitude. This is to avoid any overpotential or loss of energy efficiency due to significant differences in kinetics during full flow deployment.
During the study of the reversibility of the electronic processes of each helicene, in-depth studies of the electrokinetic parameters D and k0 of each of them were conducted for practical reasons under the conditions of cyclic voltammetry. For this purpose, CVs of isolated electronic events were performed at seven different scan rates (10, 25, 75, 100, 250, 400, and 500 mV·s-1). D determination was relying on the resolution of the Randles-Sevcik equations [Supplementary Equation 1] and k0 on the numerical application of the method developed by Nicholson et al. and Lavagnini et al. [Supplementary Equation 2][113,114].
The preliminary studies and results conducted on compounds nPrDMQA+ and nPrDMQANO2+ have already demonstrated good performances in static RFBs. Indeed, the reference carbenium with diffusion coefficients close to 1 × 10-5 cm-2·s-1 and k0 at 2 × 10-2 cm·s-1, both very balanced, enabled the creation of the first SORFB model based on a [4]helicenium[99]. In the case of nPrDMQANO2+, it was observed that the introduction of a nitro group on the DMQA+ scaffold had consequences on the potential of the electronic processes and on the value of D, and especially of k0. The electronic transfer in oxidation is five times less efficient in this case (k0Red = 3.7 × 10-2 cm·s-1, k0Ox = 0.8 × 10-2 cm·s-1), resulting in less cycling stability and robustness of the compound as published[100]. It was observed that the introduction of electron-donating groups OMe, NH2, and NMe2 at the X position of the DMQA+ scaffold had little impact on the potential of the reduction process but contributed to a shift towards less oxidizing values of the E1/2Ox phenomenon. This is reflected in balanced D parameters with values ranging between 4-6 × 10-5 cm-2·s-1 for nPrDMQANMe2+ and nPrDMQAOMe+, while nPrDMQANH2+ exhibits values similar to that of the DMQA+ model in correlation with low structural volume change. Similarly, for these three compounds, the k0 values in reduction are comparable to those of
When the time comes to modify the R1/R2 arms without substituting the core, the results become significantly more pronounced. The asymmetric species, nPr/PhDMQA+ and nPr/CH2CF3DMQA+, are strongly affected, with R2 = CH2CF3 leading to a decrease of one order of magnitude in the value of D for the reduction process, while the oxidation process maintains a value close to that observed for the previous compounds. This disparity is also evident in k0, with a reduction process sixteen times faster than in oxidation (k0Red = 1.6 × 10-2 cm·s-1, k0Ox = 0.1 × 10-2 cm·s-1); however, the fact that this phenomenon is bielectronic already eliminates this compound as a potential BRM. When R1 = R2, the D parameters behave more balanced, potentially related to the conservation of the molecule's symmetry. Thus, for
As most of the ROM's electrokinetic parameters presented here fall within the range of BRMs used in reported SORFBs, this prompts us to evaluate their robustness in cycling conditions. However, deployment in a complete in-flow RFB system is a task requiring numerous specific hardware adjustments, making it unsuitable for rapid screening of new compounds. That is why we aim to emphasize the practicality and relevance of evaluating new compounds in an H-cell. This model, described as “static RFB” in the literature[37,102], has the advantage of requiring only a small volume and low working concentration while subjecting the electroactive materials to more stressful cycling constraints. This allows rapid and efficient selection and facilitates the elimination of certain candidates within an accessible timeframe.
The system used here is based on a homemade H-cell consisting of two welded glass tubes for each compartment (Figure 4A, details in Supplementary Materials) separated by a porous frit that serves as a membrane. This design is particularly suitable for BRMs as it allows us to determine whether the molecule can tolerate the use of a porous separator without ion crossover consideration. Each “pole” of the battery is equipped with a highly conductive RVC electrode that boasts a significant specific surface area (~33 cm2 within the dimensions used in this study) and a magnetic stir bar, aiding in the diffusion of species in solution. The side where the working electrode (Ew) will be considered is also fitted with an AgNO3 reference electrode, similar to the one previously employed in the characterization of a three-electrode cell.
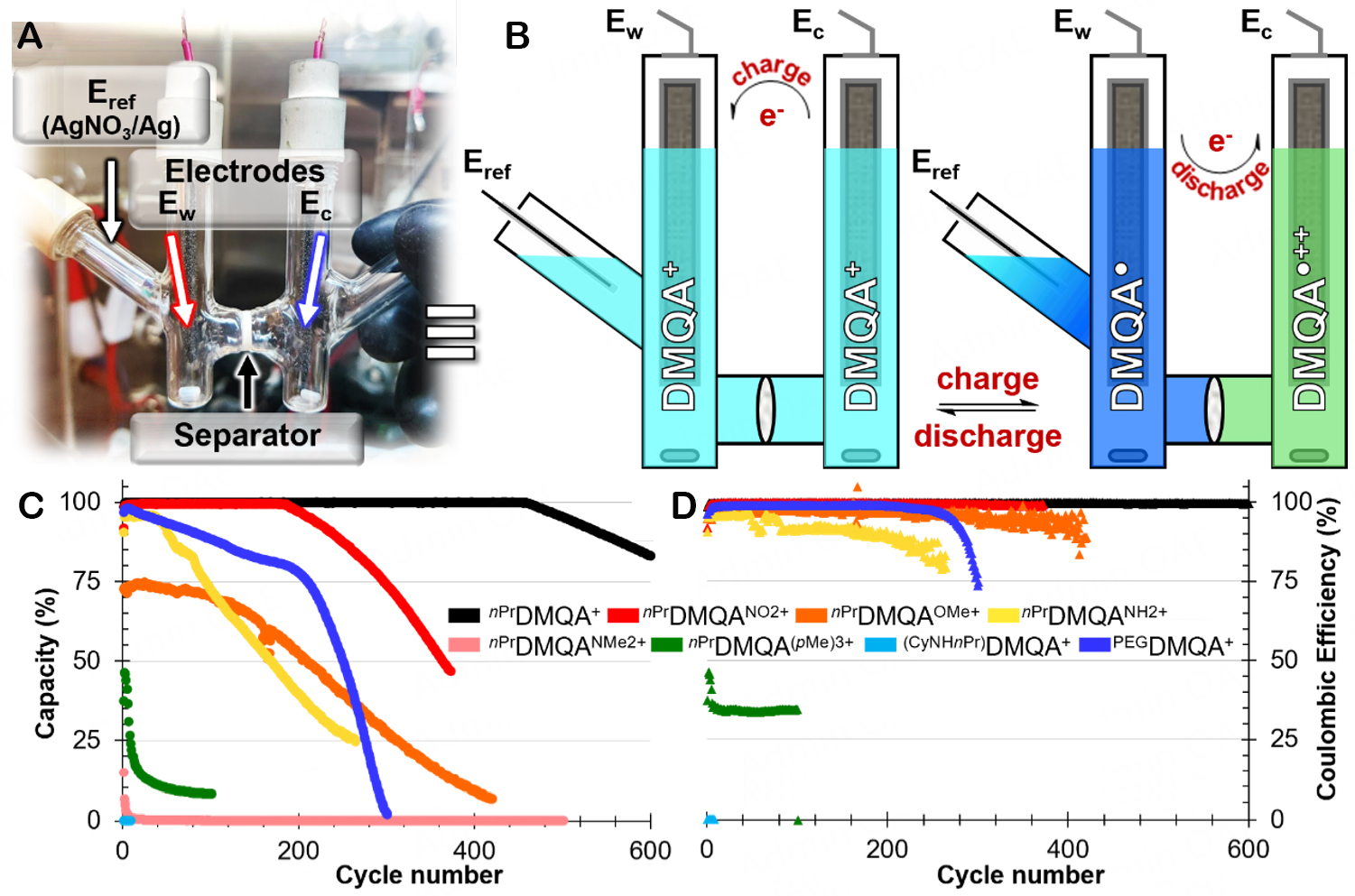
Figure 4. (A) Picture of homemade H-cell “static RFB”, full details available in Supplementary Figure 9. (B) Scheme of the H-cell in initial and in charged state (reduction occurring at Ew) loaded with 2 × 5 mL of 1 mM R1/R2DMQAX/Y+ in 0.1 M TBAPF6 CH3CN. Display of 134 μAh H-cell cycling monitoring via (C) Capacity in discharge and (D) Coulombic efficiency of nPrDMQA+, nPrDMQANO2+,
Upon the selection of [4]helicenium exhibiting electrokinetic parameters compatible with deployment for RFB, each of them is tested at a concentration of 1 mM in acetonitrile in the presence of 0.1 M TBAPF6
The cycling protocol for each of the compounds involves charging and discharging at 90% of the theoretical capacity at |5| mA. Due to its static design, this type of cell primarily relies on the diffusion of species in solution to access the entirety of the electron exchange. Hence, a constant current followed by a constant voltage (CCCV) protocol was employed. This setup was developed with a reference electrode to control the voltage at Ew, and therefore, the potential limits reached will be E1/2Red ± 300 mV. In the static RFB, the key metrics of interest are the discharge capacity (Qdis) achieved and the coulombic efficiency (CE) attained during the cycles. Thus, it is observed that nPrDMQA+ maintains a consistent Qdis = 100% until the 461st cycle, followed by a slow degradation while maintaining a near-perfect CE (Figure 4C and D, black). Similarly,
The modification of the DMQA+ core has a significant impact on the electrophysical characteristics of ROMs, as demonstrated above. However, it seems that the variation in the nature of the R1/R2 arms - when allowing the conservation of two entirely reversible electrochemical events - has little effect on the electrokinetic potentials and parameters of this BRM class. However, when the cycling of (CyNHnPr)DMQA+ is assessed, the result is disastrous. The Qdis is < 0.3%, and the CE value is null, illustrating how an evaluation in a three-electrode cell is insufficient for characterizing the suitability of a new BRM for SORFB. Thus, bulk electrolysis, such as static RFB cycling, is far too damaging for (CyNHnPr)DMQA+ (Figure 4C and D, cyan). Finally, the proposal to integrate pegyls arms seemed promising, as the CE value is > 98% and remains nearly constant up to cycle 200. Subsequently, a decay of 0.24% per cycle appears and can be correlated with what is observed in terms of Qdis values. Indeed, it is observed that PEGDMQA+ exhibits two decay regimes: the first, from cycle 5 to 200, shows its Qdis drop from 98% to 78% at a rate of 0.10% per cycle, and then, at cycle 201, this phenomenon accelerates with a decline of 0.74% per cycle (Figure 4C and D, deep blue). This behavior is not easily rationalized, but it is worth remembering that for effective discrimination, the H-cell system and the current intensities used here are extremely stressful for the electroactive material. A CE value and Qdis access rates exceeding 95% still make it a convincing BRM that deserves to be evaluated in a flow system.
CONCLUSIONS
The significance of this research work resides in the practicality of an empirical methodology, selecting an ambipolar scaffold and simple electrochemistry tools, to effectively identify and screen a variety of new BRMs. The electrochemical richness of the [4]helicenium class has illustrated how the choice of a tunable core is a rapid pathway to numerous bipolar compounds with a wide range of possible substitutions. The qualification through measurements of essential electrochemical parameters, such as diffusion coefficients and electron transfer rate constants, could be carried out using a simple three-electrode cell, making this process easily accessible. Finally, the assessment of the robustness of these BRMs in a static RFB cell constitutes a quick tool for discriminating the most promising and robust ROMs in bulk electrolysis, with the future aim of deployment in a flow RFB cell. Through the importance of this research effort, we hope to assist research groups working on the development of new BRMs in accessing promising systems more swiftly.
A potential future enhancement of this methodology could involve the incorporation of Machine Learning tools capable of predicting the characteristics of potential BRMs through computational design, based on the collection of extensive data[116,117]. This would require active participation from the community in building a large database, but it has the potential to predict the entirety of electrokinetic parameters, decomposition pathways, and potential improvements for existing bipolar molecules.
DECLARATIONS
Acknowledgments
We are thankful to Mubarak Hossain and Dr. Aslam C. Shaikh for having previously synthesized batches of nPrDMQA+, nPrDMQANO2+, nPrDMQAOMe+, nPrDMQANH2+, and nPrDMQANMe2+. Moutet J is grateful to
Authors’ contributions
Wrote the manuscript and discussed their conclusions: Moutet J, Gianetti TL
Developed, synthetized and characterized nPrDMQA(pMe)3+: El-Assaad TH
Synthetized and characterized [6]helicenium: Kaur R
Developed, synthetized and characterized nPr/CH2CF3DMQA+: Mills DD
Planned the experiments and conducted all electrochemical experiments; developed, synthetized and characterized (CyNHnPr)DMQA+: Moutet J
All authors have given approval to the final version of the manuscript.
Availability of data and materials
Nomenclature, synthesis, CVs at various scan rates, individual H-cell cycling, and additional data are available in Supplementary Materials. 1H, 13C and 19F NMR of each new compound are available in Supplementary Figures 18-35.
Financial support and sponsorship
Financial support comes from the University of Arizona, Salt River Project (Phoenix, AZ), and Research Corporation for Science Advancement Cottrell Scholarship 2021 (Award #27536). All NMR data were collected in the NMR facility of the Department of Chemistry and Biochemistry at the University of Arizona, RRID:SCR_012716. The purchase of the Bruker NEO 500 MHz spectrometer was supported by the National Science Foundation under Grant Number 1920234 and the University of Arizona.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
Supplementary Materials
REFERENCES
2. Weitemeyer S, Kleinhans D, Vogt T, Agert C. Integration of renewable energy sources in future power systems: the role of storage. Renew Energy 2015;75:14-20.
3. Gür TM. Review of electrical energy storage technologies, materials and systems: challenges and prospects for large-scale grid storage. Energy Environ Sci 2018;11:2696-767.
4. International Energy Agency. Net zero roadmap: a global pathway to keep the 1.5 °C goal in reach; Paris: IEA. 2023. Available from: https://www.iea.org/reports/net-zero-roadmap-a-global-pathway-to-keep-the-15-0c-goal-in-reach [Last accessed on 13 Mar 2024].
5. Hirsh HS, Li Y, Tan DHS, Zhang M, Zhao E, Meng YS. Sodium-ion batteries paving the way for grid energy storage. Adv Energy Mater 2020;10:2001274.
6. Shang W, Yu W, Liu Y, et al. Rechargeable alkaline zinc batteries: progress and challenges. Energy Stor Mater 2020;31:44-57.
7. Buckingham R, Asset T, Atanassov P. Aluminum-air batteries: a review of alloys, electrolytes and design. J Power Sources 2021;498:229762.
8. Huang H, Li D, Hou L, et al. Advanced protective layer design on the surface of Mg-based metal and application in batteries: challenges and progress. J Power Sources 2022;542:231755.
9. Goodenough JB, Whittingham MS, Yoshino A. The Nobel prize in chemistry 2019. For the development of lithium-ion batteries. 2019. Available from: https://www.nobelprize.org/prizes/chemistry/2019/ [Last accessed on 13 Mar 2024].
10. Masias A, Marcicki J, Paxton WA. Opportunities and challenges of lithium ion batteries in automotive applications. ACS Energy Lett 2021;6:621-30.
11. Wu J, Zhou T, Zhong B, Wang Q, Liu W, Zhou H. Designing anion-derived solid electrolyte interphase in a siloxane-based electrolyte for lithium-metal batteries. ACS Appl Mater Interfaces 2022;14:27873-81.
12. Tarascon JM, Armand M. Issues and challenges facing rechargeable lithium batteries. Nature 2001;414:359-67.
14. Yang Z, Zhang J, Kintner-Meyer MC, et al. Electrochemical energy storage for green grid. Chem Rev 2011;111:3577-613.
15. Badwal SP, Giddey SS, Munnings C, Bhatt AI, Hollenkamp AF. Emerging electrochemical energy conversion and storage technologies. Front Chem 2014;2:79.
16. Dunn B, Kamath H, Tarascon JM. Electrical energy storage for the grid: a battery of choices. Science 2011;334:928-35.
17. Alotto P, Guarnieri M, Moro F. Redox flow batteries for the storage of renewable energy: a review. Renew Sustain Energy Rev 2014;29:325-35.
18. Ravikumar MK, Rathod S, Jaiswal N, Patil S, Shukla A. The renaissance in redox flow batteries. J Solid State Electr 2017;21:2467-88.
19. Sánchez-Díez E, Ventosa E, Guarnieri M, et al. Redox flow batteries: status and perspective towards sustainable stationary energy storage. J Power Sources 2021;481:228804.
20. Zeng YK, Zhao TS, An L, Zhou XL, Wei L. A comparative study of all-vanadium and iron-chromium redox flow batteries for large-scale energy storage. J Power Sources 2015;300:438-43.
21. Suttil JA, Kucharyson JF, Escalante-garcia IL, et al. Metal acetylacetonate complexes for high energy density non-aqueous redox flow batteries. J Mater Chem A 2015;3:7929-38.
22. Beh ES, De Porcellinis D, Gracia RL, Xia KT, Gordon RG, Aziz MJ. A neutral pH aqueous organic-organometallic redox flow battery with extremely high capacity retention. ACS Energy Lett 2017;2:639-44.
23. Lourenssen K, Williams J, Ahmadpour F, Clemmer R, Tasnim S. Vanadium redox flow batteries: a comprehensive review. J Energy Stor 2019;25:100844.
24. Park M, Beh ES, Fell EM, et al. A high voltage aqueous zinc-organic hybrid flow battery. Adv Energy Mater 2019;9:1900694.
25. DOE office of ARPR-E. GRIDS program overview. Available from: https://arpa-e.energy.gov/sites/default/files/documents/files/GRIDS_ProgramOverview.pdf [Last accessed on 13 Mar 2024].
26. U.S. Department of Health and Human Services Public Health Service Agency for Toxic Substances and Disease Registry. Toxicological profile for vanadium. In: ATSDR’s toxicological profiles; Boca Raton, FL: CRC Press. 2012. Available from: https://www.atsdr.cdc.gov/toxprofiles/tp58.pdf [Last accessed on 20 Mar 2024].
27. Wittman RM, Perry ML, Lambert TN, Chalamala BR, Preger Y. Perspective - on the need for reliability and safety studies of grid-scale aqueous batteries. J Electrochem Soc 2020;167:090545.
28. Park M, Ryu J, Wang W, Cho J. Material design and engineering of next-generation flow-battery technologies. Nat Rev Mater 2017;2:16080.
29. Rhodes Z, Cabrera-pardo JR, Li M, Minteer SD. Electrochemical advances in non-aqueous redox flow batteries. Isr J Chem 2021;61:101-12.
30. Kortekaas L, Fricke S, Korshunov A, Cekic-laskovic I, Winter M, Grünebaum M. Building bridges: unifying design and development aspects for advancing non-aqueous redox-flow batteries. Batteries 2023;9:4.
31. Winsberg J, Hagemann T, Janoschka T, Hager MD, Schubert US. Redox-flow batteries: from metals to organic redox-active materials. Angew Chem Int Ed 2017;56:686-711.
32. Kim J, Kim Y, Yoo J, Kwon G, Ko Y, Kang K. Organic batteries for a greener rechargeable world. Nat Rev Mater 2023;8:54-70.
33. Ding Y, Zhang C, Zhang L, Zhou Y, Yu G. Molecular engineering of organic electroactive materials for redox flow batteries. Chem Soc Rev 2018;47:69-103.
34. Shrestha A, Hendriks KH, Sigman MS, Minteer SD, Sanford MS. Realization of an asymmetric non-aqueous redox flow battery through molecular design to minimize active species crossover and decomposition. Chemistry 2020;26:5369-73.
35. Perry ML, Saraidaridis JD, Darling RM. Crossover mitigation strategies for redox-flow batteries. Curr Opin Electrochem 2020;21:311-8.
36. Doris SE, Ward AL, Baskin A, et al. Macromolecular design strategies for preventing active-material crossover in non-aqueous all-organic redox-flow batteries. Angew Chem Int Ed 2017;56:1595-9.
37. Hendriks KH, Robinson SG, Braten MN, et al. High-performance oligomeric catholytes for effective macromolecular separation in nonaqueous redox flow batteries. ACS Cent Sci 2018;4:189-96.
38. Tsehaye MT, Mourouga G, Schmidt TJ, et al. Towards optimized membranes for aqueous organic redox flow batteries: correlation between membrane properties and cell performance. Renew Sustain Energy Rev 2023;173:113059.
39. Robb BH, George TY, Davis CM, et al. Sulfonated diels-alder poly(phenylene) membrane for efficient ion-selective transport in aqueous metalorganic and organic redox flow batteries. J Electrochem Soc 2023;170:030515.
40. Mazumder MMR, Jadhav RG, Minteer SD. Phenyl acrylate-based cross-linked anion exchange membranes for non-aqueous redox flow batteries. ACS Mater Au 2023;3:557-68.
41. Navalpotro P, Sierra N, Trujillo C, Montes I, Palma J, Marcilla R. Exploring the versatility of membrane-free battery concept using different combinations of immiscible redox electrolytes. ACS Appl Mater Interfaces 2018;10:41246-56.
42. Potash RA, Mckone JR, Conte S, Abruña HD. On the benefits of a symmetric redox flow battery. J Electrochem Soc 2016;163:A338-44.
43. Janoschka T, Friebe C, Hager MD, Martin N, Schubert US. An approach toward replacing vanadium: a single organic molecule for the anode and cathode of an aqueous redox-flow battery. ChemistryOpen 2017;6:216-20.
44. Chen R. Redox flow batteries: mitigating cross-contamination via bipolar redox-active materials and bipolar membranes. Curr Opin Electrochem 2023;37:101188.
45. Li M, Case J, Minteer SD. Bipolar redox-active molecules in non-aqueous organic redox flow batteries: status and challenges. ChemElectroChem 2021;8:1215-32.
46. Kosswattaarachchi AM, Friedman AE, Cook TR. Characterization of a BODIPY dye as an active species for redox flow batteries. ChemSusChem 2016;9:3317-23.
47. Ma T, Pan Z, Miao L, et al. Porphyrin-based symmetric redox-flow batteries towards cold-climate energy storage. Angew Chem 2018;130:3212-6.
48. Geysens P, Li Y, Vankelecom I, Fransaer J, Binnemans K. Highly soluble 1,4-diaminoanthraquinone derivative for nonaqueous symmetric redox flow batteries. ACS Sustain Chem Eng 2020;8:3832-43.
49. Tracy JS, Horst ES, Roytman VA, Toste FD. Development of high-voltage bipolar redox-active organic molecules through the electronic coupling of catholyte and anolyte structures. Chem Sci 2022;13:10806-14.
50. Liu Y, Dai G, Chen Y, et al. Effective design strategy of small bipolar molecules through fused conjugation toward 2.5 V based redox flow batteries. ACS Energy Lett 2022;7:1274-83.
51. Steen JS, Nuismer JL, Eiva V, et al. Blatter radicals as bipolar materials for symmetrical redox-flow batteries. J Am Chem Soc 2022;144:5051-8.
52. Hagemann T, Winsberg J, Häupler B, et al. A bipolar nitronyl nitroxide small molecule for an all-organic symmetric redox-flow battery. NPG Asia Mater 2017;9:e340.
53. Hwang S, Kim H, Ryu JH, Oh SM. N-ferrocenylphthalimide; A single redox couple formed by attaching a ferrocene moiety to phthalimide for non-aqueous flow batteries. J Power Sources 2018;395:60-5.
54. Friedl J, Lebedeva MA, Porfyrakis K, Stimming U, Chamberlain TW. All-fullerene-based cells for nonaqueous redox flow batteries. J Am Chem Soc 2018;140:401-5.
55. Hwang S, Kim H, Ryu JH, Oh SM. N-(α-ferrocenyl)ethylphthalimide as a single redox couple for non-aqueous flow batteries. J Power Sources 2019;421:1-5.
56. Zhen Y, Zhang C, Yuan J, Zhao Y, Li Y. Ferrocene/anthraquinone based bi-redox molecule for symmetric nonaqueous redox flow battery. J Power Sources 2020;480:229132.
57. Xu D, Zhang C, Zhen Y, Li Y. Ferrocene/phthalimide ionic bipolar redox-active molecule for symmetric nonaqueous redox flow batteries. ACS Appl Energy Mater 2021;4:8045-51.
58. Liu B, Tang CW, Sheong FK, Jia G, Zhao T. Artificial bipolar redox-active molecule for symmetric nonaqueous redox flow batteries. ACS Sustain Chem Eng 2022;10:613-21.
59. Nambafu GS, Delmo EP, Bin Shahid U, et al. Pyromellitic diimide based bipolar molecule for total organic symmetric redox flow battery. Nano Energy 2022;94:106963.
60. Etkind SI, Lopez J, Zhu YG, et al. Thianthrene-based bipolar redox-active molecules toward symmetric all-organic batteries. ACS Sustain Chem Eng 2022;10:11739-50.
61. Duan W, Vemuri RS, Milshtein JD, et al. A symmetric organic-based nonaqueous redox flow battery and its state of charge diagnostics by FTIR. J Mater Chem A 2016;4:5448-56.
62. Charlton GD, Barbon SM, Gilroy JB, Dyker CA. A bipolar verdazyl radical for a symmetric all-organic redox flow-type battery. J Energy Chem 2019;34:52-6.
63. Armstrong CG, Hogue RW, Toghill KE. Application of the dianion croconate violet for symmetric organic non-aqueous redox flow battery electrolytes. J Power Sources 2019;440:227037.
64. Raihan M, Dyker CA. Ester-substituted bispyridinylidenes: double concerted two-electron bipolar molecules for symmetric organic redox flow batteries. ACS Energy Lett 2023;8:3314-22.
65. Winsberg J, Stolze C, Muench S, Liedl F, Hager MD, Schubert US. TEMPO/phenazine combi-molecule: a redox-active material for symmetric aqueous redox-flow batteries. ACS Energy Lett 2016;1:976-80.
66. Dmello R, Milshtein JD, Brushett FR, Smith KC. Cost-driven materials selection criteria for redox flow battery electrolytes. J Power Sources 2016;330:261-72.
67. Sentyurin VV, Levitskiy OA, Magdesieva TV. Molecular design of ambipolar redox-active molecules II: closed-shell systems. Curr Opin Electrochem 2020;24:6-14.
68. Sentyurin VV, Levitskiy OA, Magdesieva TV. Molecular design of ambipolar redox-active open-shell molecules: principles and implementations. Curr Opin Electrochem 2020;24:15-23.
69. Steen JS, de Vries F, Hjelm J, Otten E. Bipolar verdazyl radicals for symmetrical batteries: properties and stability in all states of charge. Chemphyschem 2023;24:e202200779.
70. Broere DL, Plessius R, van der Vlugt JI. New avenues for ligand-mediated processes--expanding metal reactivity by the use of redox-active catechol, o-aminophenol and o-phenylenediamine ligands. Chem Soc Rev 2015;44:6886-915.
72. Laursen BW, Krebs FC, Nielsen MF, Bechgaard K, Christensen JB, Harrit N. 2,6,10-Tris(dialkylamino)trioxatriangulenium ions. Synthesis, structure, and properties of exceptionally stable carbenium ions. J Am Chem Soc 1998;120:12255-63.
73. Nicolas C, Lacour J. Triazatriangulenium cations: highly stable carbocations for phase-transfer catalysis. Org Lett 2006;8:4343-6.
74. Goodman H, Mei L, Gianetti TL. Molecular orbital insights of transition metal-stabilized carbocations. Front Chem 2019;7:365.
75. Wilkins LC, Kim Y, Litle ED, Gabbaï FP. Stabilized carbenium ions as latent, Z-type ligands. Angew Chem Int Ed 2019;131:18434-8.
76. Mei L, Veleta JM, Bloch J, et al. Tunable carbocation-based redox active ambiphilic ligands: synthesis, coordination and characterization. Dalton Trans 2020;49:16095-105.
77. Litle ED, Wilkins LC, Gabbaï FP. Ligand-enforced intimacy between a gold cation and a carbenium ion: impact on stability and reactivity. Chem Sci 2021;12:3929-36.
78. Karimi M, Borthakur R, Dorsey CL, Chen CH, Lajeune S, Gabbaï FP. Bifunctional carbenium dications as metal-free catalysts for the reduction of oxygen. J Am Chem Soc 2020;142:13651-6.
79. Shaikh AC, Veleta JM, Moutet J, Gianetti TL. Trioxatriangulenium (TOTA+) as a robust carbon-based Lewis acid in frustrated Lewis pair chemistry. Chem Sci 2021;12:4841-9.
80. Mei L, Veleta JM, Gianetti TL. Helical carbenium ion: a versatile organic photoredox catalyst for red-light-mediated reactions. J Am Chem Soc 2020;142:12056-61.
81. Mei L, Gianetti T. Helical carbenium ion-based organic photoredox catalyst: a versatile and sustainable option in red-light-induced reactions. Synlett 2021;32:337-4.
82. Mei L, Moutet J, Stull SM, Gianetti TL. Synthesis of CF3-containing spirocyclic indolines via a red-light-mediated trifluoromethylation/dearomatization cascade. J Org Chem 2021;86:10640-53.
83. Hossain MM, Shaikh AC, Moutet J, Gianetti TL. Photocatalytic α-arylation of cyclic ketones. Nat Synth 2022;1:147-57.
84. Nowack MH, Moutet J, Laursen BW, Gianetti TL. Triangulenium ions: versatile organic photoredox catalysts for green-light-mediated reactions. Synlett 2024;35:307-12.
85. Singh PP, Singh J, Srivastava V. Visible-light acridinium-based organophotoredox catalysis in late-stage synthetic applications. RSC Adv 2023;13:10958-86.
86. Žurauskas J, Boháčová S, Wu S, et al. Electron-poor acridones and acridiniums as super photooxidants in molecular photoelectrochemistry by unusual mechanisms. Angew Chem Int Ed 2023;62:e202307550.
87. Moutet J, Nowack MH, Mills DD, Lozier DL, Laursen BW, Gianetti TL. Planar carbenium ions for robust symmetrical all organic redox flow batteries. Mater Adv 2023;4:4598-606.
88. Bosson J, Bisballe N, Laursen BW, Lacour J. Chapter 4: cationic triarylcarbenium helicenes: synthesis, resolution, and applications. Weinheim, Germany: Wiley; 2022. pp. 127-65.
89. Herse C, Bas D, Krebs FC, et al. A highly configurationally stable [4]heterohelicenium cation. Angew Chem Int Ed 2003;42:3162-6.
90. Laleu B, Mobian P, Herse C, et al. Resolution of [4]heterohelicenium dyes with unprecedented Pummerer-like chemistry. Angew Chem Int Ed 2005;44:1879-83.
91. Kel O, Sherin P, Mehanna N, Laleu B, Lacour J, Vauthey E. Excited-state properties of chiral [4]helicene cations. Photochem Photobiol Sci 2012;11:623-31.
92. Bosson J, Gouin J, Lacour J. Cationic triangulenes and helicenes: synthesis, chemical stability, optical properties and extended applications of these unusual dyes. Chem Soc Rev 2014;43:2824-40.
93. Wallabregue A, Sherin P, Guin J, Besnard C, Vauthey E, Lacour J. Modular synthesis of pH-sensitive fluorescent diaza[4]helicenes. Eur J Org Chem 2014;2014:6431-8.
94. Delgado IH, Pascal S, Wallabregue A, et al. Functionalized cationic [4]helicenes with unique tuning of absorption, fluorescence and chiroptical properties up to the far-red range. Chem Sci 2016;7:4685-93.
95. Li H, Voci S, Wallabregue A, et al. Efficient annihilation electrochemiluminescence of cationic helicene luminophores. ChemElectroChem 2017;4:1750-6.
96. Tarrieu R, Delgado IH, Zinna F, et al. Hybrids of cationic [4]helicene and N-heterocyclic carbene as ligands for complexes exhibiting (chir)optical properties in the far red spectral window. Chem Commun 2021;57:3793-6.
97. Sørensen TJ, Nielsen MF, Laursen BW. Synthesis and Stability of N,N′-Dialkyl-1,13-dimethoxyquinacridinium (DMQA+): a [4]helicene with multiple redox states. ChemPlusChem 2014;79:1030-5.
98. Shaikh AC, Moutet J, Veleta JM, et al. Persistent, highly localized, and tunable [4]helicene radicals. Chem Sci 2020;11:11060-7.
99. Moutet J, Veleta JM, Gianetti TL. Symmetric, robust, and high-voltage organic redox flow battery model based on a helical carbenium ion electrolyte. ACS Appl Energy Mater 2021;4:9-14.
100. Moutet J, Mills D, Hossain MM, Gianetti TL. Increased performance of an all-organic redox flow battery model via nitration of the [4]helicenium DMQA ion electrolyte. Mater Adv 2022;3:216-23.
101. Yan Y, Robinson SG, Sigman MS, Sanford MS. Mechanism-based design of a high-potential catholyte enables a 3.2 V all-organic nonaqueous redox flow battery. J Am Chem Soc 2019;141:15301-6.
102. Antoni PW, Bruckhoff T, Hansmann MM. Organic redox systems based on pyridinium-carbene hybrids. J Am Chem Soc 2019;141:9701-11.
103. Mobian P, Nicolas C, Francotte E, Bürgi T, Lacour J. Synthesis, resolution, and VCD analysis of an enantiopure diazaoxatricornan derivative. J Am Chem Soc 2008;130:6507-14.
104. Gong K, Fang Q, Gu S, Li SFY, Yan Y. Nonaqueous redox-flow batteries: organic solvents, supporting electrolytes, and redox pairs. Energy Environ Sci 2015;8:3515-30.
105. Korshunov A, Milner MJ, Grünebaum M, Studer A, Winter M, Cekic-laskovic I. An oxo-verdazyl radical for a symmetrical non-aqueous redox flow battery. J Mater Chem A 2020;8:22280-91.
106. Laursen BW, Sørensen TJ. Synthesis of super stable triangulenium dye. J Org Chem 2009;74:3183-5.
107. Bisballe N, Laursen BW. What is best strategy for water soluble fluorescence dyes? A case study using long fluorescence lifetime DAOTA dyes. Chemistry 2020;26:15969-76.
108. Moutet J, Mills DD, Lozier DL, Gianetti TL. [4]helicenium ion as bipolar redox material for symmetrical fully organic pole-less redox flow battery. Batteries Supercaps 2024:e202300519.
109. Helicenes: synthesis, properties and applications. Crassous J, Stará IG, Starý I, editors. Weinheim, Germany: Wiley; 2022. pp. 1-542.
110. Torricelli F, Bosson J, Besnard C, Chekini M, Bürgi T, Lacour J. Modular synthesis, orthogonal post-functionalization, absorption, and chiroptical properties of cationic [6]helicenes. Angew Chem Int Ed 2013;52:1796-800.
111. Yao Y, Lei J, Shi Y, Ai F, Lu Y. Assessment methods and performance metrics for redox flow batteries. Nat Energy 2021;6:582-8.
112. Wang H, Sayed SY, Luber EJ, et al. Redox flow batteries: how to determine electrochemical kinetic parameters. ACS Nano 2020;14:2575-84.
113. Nicholson RS. Theory and application of cyclic voltammetry for measurement of electrode reaction kinetics. Anal Chem 1965;37:1351-5.
114. Lavagnini I, Antiochia R, Magno F. An extended method for the practical evaluation of the standard rate constant from cyclic voltammetric data. Electroanalysis 2004;16:505-6.
115. Cavallotti C, Derudi M, Rota R. On the mechanism of decomposition of the benzyl radical. Proc Combust Inst 2009;32:115-21.
116. Yao Z, Lum Y, Johnston A, et al. Machine learning for a sustainable energy future. Nat Rev Mater 2023;8:202-15.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Moutet J, El-Assaad TH, Kaur R, Mills DD, Gianetti TL. Designing the next generation of symmetrical organic redox flow batteries using helical carbocations. Energy Mater 2024;4:400024. http://dx.doi.org/10.20517/energymater.2023.92
AMA Style
Moutet J, El-Assaad TH, Kaur R, Mills DD, Gianetti TL. Designing the next generation of symmetrical organic redox flow batteries using helical carbocations. Energy Materials. 2024; 4(3): 400024. http://dx.doi.org/10.20517/energymater.2023.92
Chicago/Turabian Style
Moutet, Jules, Tarek H. El-Assaad, Ramandeep Kaur, David D. Mills, Thomas L. Gianetti. 2024. "Designing the next generation of symmetrical organic redox flow batteries using helical carbocations" Energy Materials. 4, no.3: 400024. http://dx.doi.org/10.20517/energymater.2023.92
ACS Style
Moutet, J.; El-Assaad TH.; Kaur R.; Mills DD.; Gianetti TL. Designing the next generation of symmetrical organic redox flow batteries using helical carbocations. Energy Mater. 2024, 4, 400024. http://dx.doi.org/10.20517/energymater.2023.92
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 2 clicks
Cite This Article 2 clicks

 Like This Article 5
likes
Like This Article 5
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.